
Abstract
Portopulmonary hypertension (PoPH) is defined by the combination of portal hypertension and precapillary pulmonary arterial hypertension (PAH). Very little is known about this process in pediatric patients but prognosis is generally poor. We review our institutional experience and report on five patients with pediatric PoPH. The median age of PoPH diagnosis was six years and PAH was 14 years. PAH diagnosis was made by echocardiogram in all patients, four of whom also had cardiac catheterization. The median mean pulmonary artery pressure (mPAP) was 48.5 mmHg (interquartile range [IQR] = 46–60) with a median pulmonary vascular resistance index (PVRi) of 9 WU*M2 (IQR = 8–22). All were acute pulmonary vasodilator testing non-responsive. All patients received targeted therapies. Three of five patients (60%) died despite an evidence-based approach to care. Of those who died, timing from the PoPH diagnosis to death ranged from three days to three years. Based upon our limited experience, PoPH is a disorder with significant mortality in childhood and challenges in treatment. Future research, focused on screening and early targeted treatment strategies, may alter the current dismal prognosis for these children.
Keywords
Introduction
Traditionally, disease processes that affect either the liver or the lungs are distinct, but several notable exceptions have challenged physicians. Mantz and Craig first reported one such relationship in 1951, and following several decades the condition has been classified as portopulmonary hypertension. 1 Portopulmonary hypertension (PoPH) is characterized by the combination of portal hypertension and pulmonary arterial hypertension (PAH) (mean pulmonary artery pressure [mPAP] > 25 mmHg, pulmonary capillary wedge pressure [PCWP] < 15 mmHg, and pulmonary vascular resistance [PVR] > 3 Wood units [WU]).
On histopathology, the pulmonary vascular pathology observed in PoPH is identical to idiopathic pulmonary arterial hypertension (IPAH). Findings include vasoconstriction, endothelial and smooth muscle proliferation, plexogenic arteriopathy, in situ thrombosis, and fibrosis. 2 In adults with liver cirrhosis, the incidence of PoPH is reported to be in the range of 2–8%.3,4 Data from the Registry to Evaluate Early and Longterm PAH Disease Management (REVEAL) registry showed that patients with PoPH had poorer survival rates and all-cause hospitalization rates when compared with patients with IPAH. 5 A diagnosis of PoPH in a cirrhosis patient is associated with high mortality after liver transplantation and may be a contraindication to transplantation.6–8
The vast majority of published studies are in adults. Very little is known about this disease process in pediatric patients. Nevertheless, in an autopsy series of children diagnosed with portal hypertension, 5.4% had histologic evidence of PAH. 9 What is known about prognosis and outcomes is limited to case reports or small case series. In order to increase the knowledge of this diagnosis in pediatrics and allow further characterization of presentation, pathophysiology, prognosis, and potential therapeutic measures, we reviewed our institutional experience with PoPH in pediatric patients. Although we acknowledge the limited generalizability and inherent bias with studying a small sample size, given the limited data available, we felt that our experience would add important information to the current knowledge base and stimulate further research.
Methods
We report five pediatric patients with a diagnosis of PoPH treated at the Pulmonary Hypertension Center at Columbia University Medical Center. A retrospective chart review of these patients was conducted. Data including demographics, clinical characteristics, cardiac and hepatic function indices, and management strategies were collected. Review of patient records was approved by the institutional review board and was exempt from written consent.
Case summaries
Patient demographics.
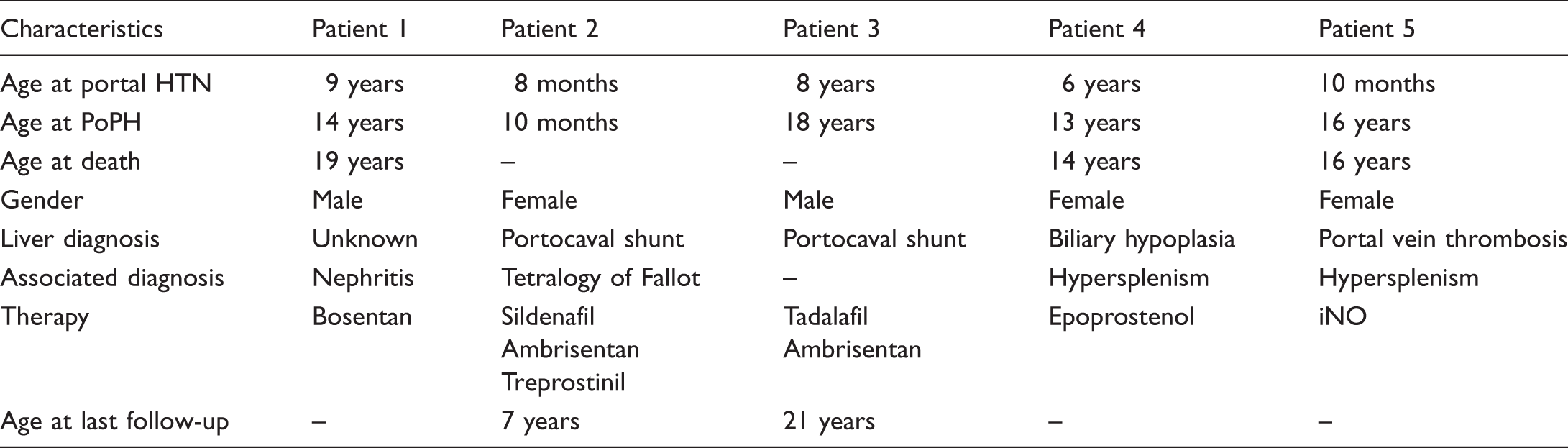
HTN, hypertension; PoPH, portopulmonary hypertension; iNO, inhaled nitric oxide.
Clinical evaluation of PoPH patients at diagnosis of PH.
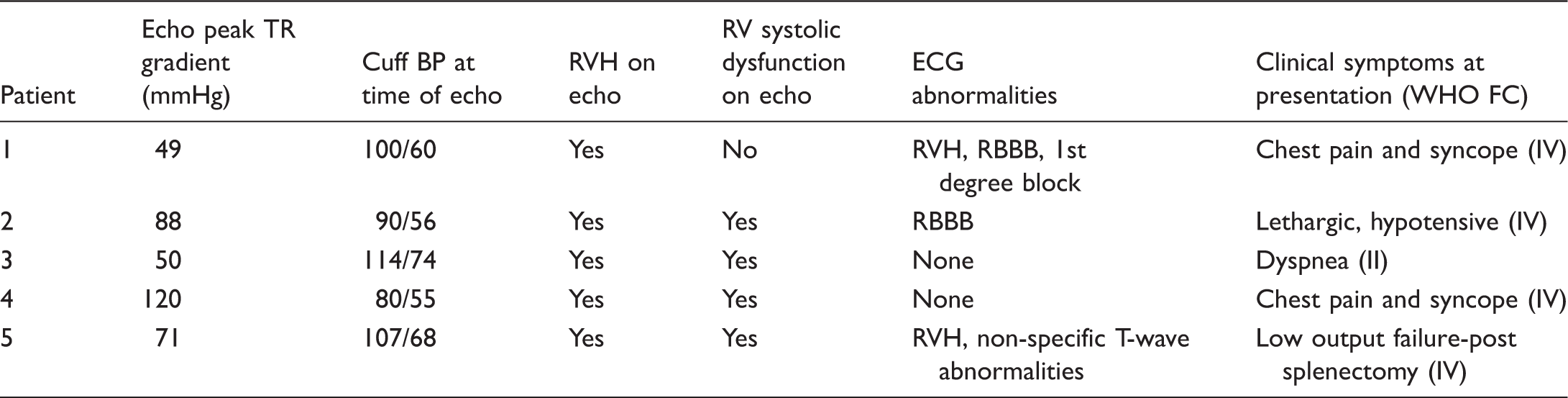
TR, tricuspid regurgitation; RVH, right ventricular hypertrophy; RV, right ventricle; ECG, electrocardiogram; RBBB, right bundle branch block.
Cardiac catheterization data.
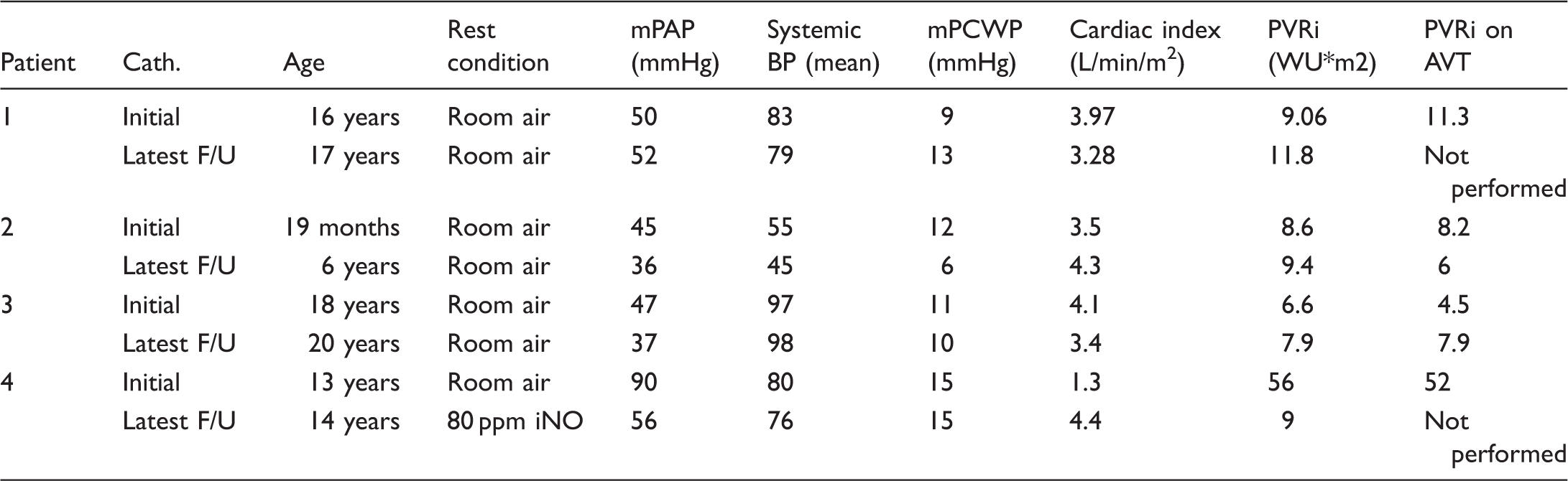
Cath., catheterization; mPAP, mean pulmonary artery pressure; BP, blood pressure; mPCWP, mean pulmonary capillary wedge pressure; PVRi, pulmonary vascular resistance index; AVT, acute pulmonary vasodilator testing; F/U, follow-up; iNO, inhaled nitric oxide.
Patient 2 is a Caucasian girl with tetralogy of Fallot, with typical anatomy, a malalignment ventricular septal defect, infundibular and valvar PS, and mildly hypoplastic branch pulmonary arteries. She was repaired at five months of age in Europe. At nine months, she presented with lethargy and was found to have supra-systemic right ventricular pressures on echocardiogram and investigation revealed a congenital portocaval shunt. She had device closure of the portocaval shunt performed in the catheterization lab but showed no improvement in her right ventricular pressures post procedure. Targeted PAH therapy was initiated at ten months of age. She was initially started on sildenafil and bosentan. She moved to the United States and was referred to our PH center at 18 months of age. She underwent cardiac catheterization that revealed mPAP of 45 mmHg and PVR index (PVRi) of 8.6 WU*M2. She was unresponsive to oxygen or inhaled nitric oxide (iNO) (Table 3). Several days after the catheterization, she presented to the emergency department with hematemesis and subsequently was diagnosed with esophageal varices. Liver biopsy findings were consistent with chronic right-sided heart failure (Table 2). She was initiated on subcutaneous treprostinil and continuous oxygen via nasal cannula. The subcutaneous treprostinil was transitioned to intravenous infusion at three years of age secondary to site issues. To address chronic thrombocytopenia from hypersplenism, initially diagnosed at the age of three years, she underwent splenic embolization at the age of six years. The period following the splenic embolization required very close coordination of care because of the tremendous fluid shifts and hemodynamic instability. She developed high output heart failure and her intravenous prostanoid dose had to be rapidly reduced while balancing her fluids and cardiorespiratory support. She is currently alive with WHO FC III symptoms on oral sildenafil and intravenous treprostinil.
Patient 3 presented at 18 years of age to our liver service for a second opinion after being diagnosed with extrahepatic congenital portocaval shunt Type IIa (Abernathy malformation) at eight years of age. He also had a history of moderate persistent asthma treated by pulmonologists on intermittent inhaled steroids and bronchodilator therapy. At referral to Cardiology, he had symptoms of easy fatigability on walking up a flight of stairs (WHO FC II–III) and desaturations following an asthma exacerbation. A prior echocardiogram at ten years of age had shown normal right ventricular systolic pressures. At 18 years, echocardiogram revealed over half systemic right ventricular pressures, mildly dilated right atrium and right ventricle with mild right ventricular dysfunction, and significant pulmonary artery dilatation. He was initially started on sildenafil, then changed to tadalafil for better compliance. He had a liver biopsy performed which revealed no evidence of fibrosis, mild centrilobar dilatation, and a normal liver architecture. At age 19, he underwent closure of his portal caval connection with placement of a covered inferior vena cava stent (32 × 39 mm Zenith Flex stent graft). Although he suffered from intestinal angina following the procedure, no bowel was lost and symptoms subsided without intervention. He currently continues on tadalafil with WHO FC II symptoms and less than half systemic PAPs on catheterization.
Patient 4 presented at the age of 13 years with a prolonged seizure and respiratory arrest requiring brief cardiopulmonary resuscitation (WHO FC IV). She had a history of congenital biliary hypoplasia and portal hypertension. Further history revealed complaints of episodic chest tightness, dyspnea, and palpitations. Chest radiograph revealed enlarged central pulmonary arteries and pruned appearance to the pulmonary vascular markings. Physical exam revealed signs of right heart failure with cold extremities and tachycardia, right ventricular heave, and a tricuspid regurgitation murmur. Echocardiogram confirmed supra-systemic right ventricular pressures and severe right ventricular dysfunction. Catheterization confirmed severe PAH, unresponsive to acute pulmonary vasodilator testing (Table 3). Subsequently, she was treated with iNO, intravenous epoprostenol, and diuretics with acute improvement over two weeks permitting discharge home. While undergoing therapy, she had marked improvement of her right heart pressures as seen on serial echocardiograms (Fig. 1). Several months later, she was readmitted with cholecystitis and sepsis. In the setting of this acute abdominal process, she had an acute worsening of her PAH and clinical decompensation requiring extracorporeal membrane oxygenation (ECMO) and subsequent balloon atrial septostomy. Despite these efforts, she died secondary to a severe pulmonary hypertensive crisis and severe sepsis.
Patient 4 echocardiogram still frame (apical view) demonstrating right heart dilation and posterior bowing of the ventricular septum at the time of diagnosis (a). (b) Significant improvement after several months of therapy.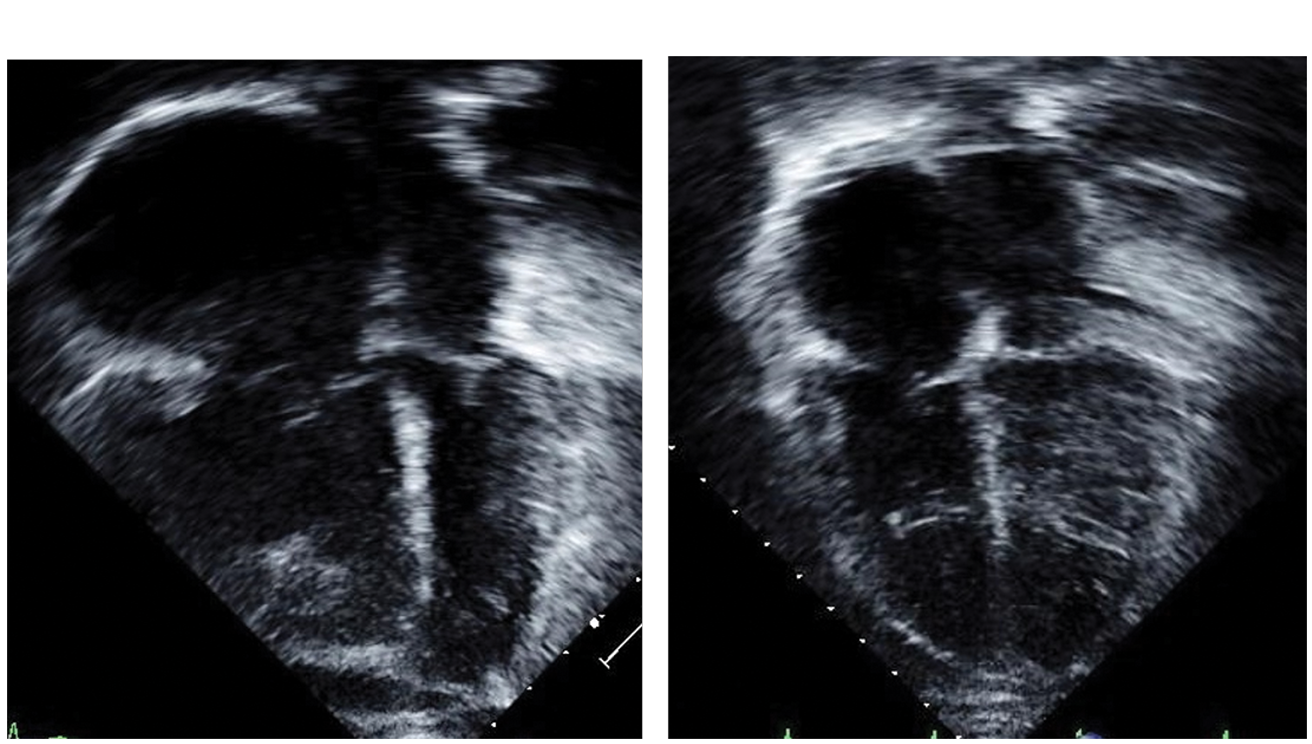
Patient 5 was a 16-year-old girl with a history of portal vein thrombosis complicated by cavernous transformation, splenomegaly with infarcts, and esophageal varices treated with multiple rounds of sclerotherapy at an outside institution. She underwent a Rex shunt (mesenteric vein to right portal vein) portal vein bypass graft and splenectomy and was admitted to our unit for postoperative monitoring. Due to persistent desaturations, tachycardia, and hypotension on postoperative day 3, an echocardiogram was performed that showed severe PAH (80% systemic) and she was started on iNO and milrinone. She initially improved on iNO at 20 parts per million. A computerized tomography scan ruled out pulmonary embolism but suggested chronic PAH. She had a sudden hypotensive arrest on postoperative day 5 and was emergently cannulated to ECMO. She had an uncontrollable abdominal hemorrhage secondary to a liver laceration (found postmortem), and low platelets from hypersplenism leading to death in a few hours after ECMO. Postmortem autopsy examination of the lung vasculature showed histopathologic changes consistent with severe PH with Heath Edwards Stage V changes in the vasculature (Fig. 2) and marked right ventricular dilation and hypertrophy consistent with chronically increased right sided pressures.
Patient 5 lung histopathology showing plexiform lesions, medial hypertrophy, and intimal hyperplasia consistent with severe PAH (Heath and Edwards Grade V/VI).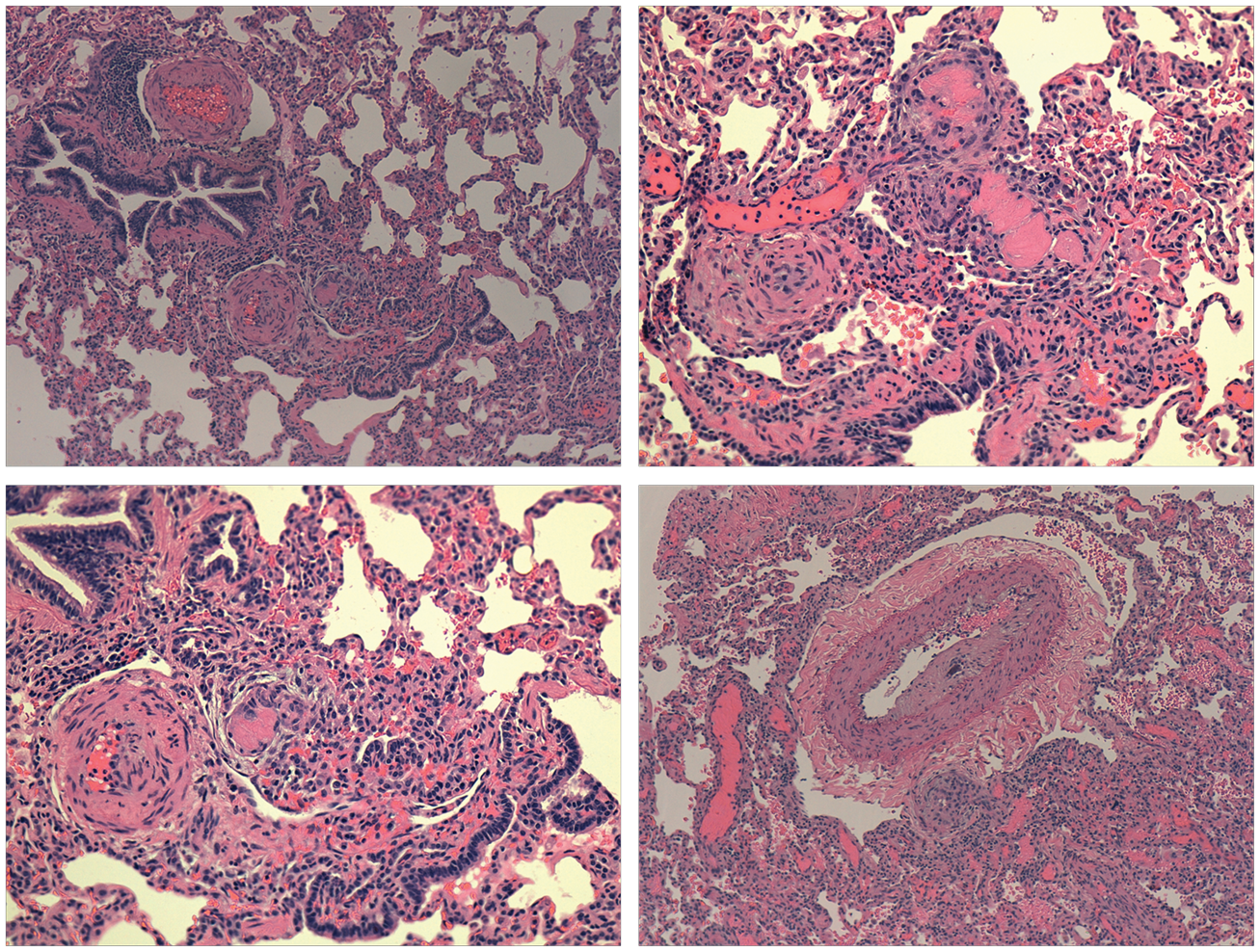
Results
Baseline patient demographic data are displayed in Table 1. Five children (three girls, two boys) were identified with PoPH. The median age of diagnosis of PoPH was six years (range = 10 months–9 years). The median age of diagnosis of PAH was 14 years (range = 8 months–18 years). The median time interval from the diagnosis of portal hypertension to the diagnosis of PAH was five years (range = 2 months–15 years). One patient had congenital heart disease (Tetralogy of Fallot). Hepatic diagnoses were varied. Patient 1 presented initially with esophageal varices and portal hypertension with an unknown underlying liver pathologic diagnosis. Two of the patients had portocaval shunts, one had underlying liver cirrhosis from biliary hypoplasia, and one had a history of portal vein thrombosis. Two patients presented with chest pain and syncope, one with lethargy, one with acute postoperative deterioration, and one patient presented with dyspnea on exertion (Table 2). The diagnosis of PAH was made by echocardiogram in all, four of whom also had cardiac catheterization. Cardiac catheterization data are summarized in Table 3. The median mPAP was 48.5 mmHg (IQR = 46–60) with a median PVRi of 9 WUm2 (IQR = 8–22). All were acute pulmonary vasodilator testing non-responsive. All patients received targeted therapies, with two on parenteral prostanoids. Three of our five patients (60%) died despite use of standard evidence-based treatment strategies utilized in pediatric PAH (one at a community hospital, one secondary to a surgical hemorrhage, and the third from fulminant sepsis, cholecystitis, and inflammation-related PH crisis). Of those who died, timing from the diagnosis of PoPH to death ranged from three days to three years.
Discussion
PoPH is a disorder with high mortality in childhood. Based upon our limited experience, disease progression can be rapid. It is likely that PoPH is under-recognized in children and hence they present often when they are very symptomatic from their PAH as was seen in our patients. The delayed diagnosis possibly contributes to the early high mortality described. The current literature is limited to small case series. Condino et al., in their experience with seven patients, report a high mortality (4/7 deaths). 10 Syncope was the most frequent presenting symptom and the patients had variable response to therapy. In a retrospective study by Kawut et al., comparing 13 adult patients with PoPH and 33 patients with IPAH, the PoPH patients had an almost threefold higher risk of death than the IPAH patients. 11
As expected, the liver pathology in our pediatric population is different from what is reported in the adult literature. In several large adult studies, alcoholic liver disease and/or hepatitis with cirrhosis are the most common etiologies of liver disease. 12 In pediatrics, congenital etiologies of liver cirrhosis are more common (i.e. biliary atresia). It is known, though, that PoPH can be seen without the intrinsic liver disease and can be seen in disease processes that cause portal hypertension without cirrhosis (portosystemic shunts). 13 Our series included two children with portosystemic shunts, consistent with the theory that PoPH is associated with portal hypertension and not necessarily intrinsic liver pathology.
It is theorized that the hyperdynamic circulatory state of portal hypertension contributes to development of both the hepatopulmonary syndrome and PoPH. The exact mechanistic link between portal hypertension and the development of pulmonary vascular disease (PVD) has not been proven. Portosystemic shunts and hepatic failure are known to lead to an increase in circulating systemic vasodilators. 14 This condition leads to splanchnic vasodilation and low systemic vascular resistance.14,15 Volume overload onto the splanchnic vasculature leads to bowel wall congestion and potentially the release of endotoxins and cytokines. 16 In addition, higher cardiac output and increased flow within the pulmonary vasculature creates increased shear forces and increased mPAPs without a resultant increase in PVR.
The pulmonary vasculature’s response to the hyperdynamic circulatory syndrome is variable. A vasodilatory response may lead to the development of hepatopulmonary syndrome. Vasoconstriction may lead to development of PVD similar to precapillary PH. 16 There may be an individual genetic or environmental predisposing factor that promotes the development of vasodilation and hepatopulmonary syndrome versus vasoconstriction and PoPH. Others have speculated that hepatopulmonary syndrome can precede the development of PoPH. Ecochard et al. recently reported six pediatric patients who had hypoxemia secondary to hepaopulmonary syndrome prior to the development of PoPH. 17
In both pediatric and adult PoPH patients, histopathologic changes are identical to IPAH.2,9,18 The pathogenesis of PAH involves a dysregulation of vasodilators and vasoconstrictors including prostacyclin, nitric oxide, endothelin-1, thromboxane A2, and serotonin.19,20 Endothelin 1, a peptide known to cause vasoconstriction, is found in increased levels in animal models of cirrhosis. 21 It has been postulated that portal hypertension causes an inflammatory response secondary to the volume overload of the splanchnic vasculature, allowing for humoral mediators, cytokines, and endotoxins to be released into the circulation 16 similar to the inflammatory process in IPAH, with elevations in interleukin-1β and interleukin-6. 22 With the release of these inflammatory mediators, there is continued increase to pulmonary blood flow, sheer stresses upon the pulmonary vasculature and subsequent vasoconstriction, and pulmonary vascular remodeling. Potentially, in portal hypertension obligate portosystemic shunting prevents metabolism of these inflammatory substances by the liver and directly affects the pulmonary vasculature. 23 This may also lead to pulmonary arteriovenous malformations in some cases.
One might expect to observe more pronounced PoPH in proportion to the severity of the liver disease. However, Benjaminov et al. demonstrated no association with the severity of liver disease and severity of PAH (based upon right heart hemodynamic measurements) in an adult study involving 62 patients with liver cirrhosis and ascites. 23
In our series, the majority of patients had severe PAH at cardiac presentation. Of the four patients who underwent cardiac catheterization, none showed reactivity to vasodilators and so calcium channel blockers were not used as therapy. Treatment was initiated in all patients upon diagnosis and therapies were selected based upon disease severity. Two patients showed long-term response with lower pulmonary pressures on parenteral prostanoids. Patient 2 continues to be treated with intravenous treprostinil. Although Patient 4 had an initial successful response to therapy, she clinical decompensated in the setting of cholecystitis, sepsis, and pulmonary hypertensive crisis leading to her demise. The other surviving patient had interventional occlusion of his portocaval shunt with improvement in his PAPs and clinical symptoms with stable oral therapy. It is evident from our small group that multiple factors including sepsis, inflammation, and hypersplenism play a role in the mortality in this group of patients.
The most effective therapy for this disease process has not been established. The use of prostanoids, calcium channel blockers, endothelin receptor antagonists, and phosphodiesterase-5 (PDE5) inhibitors have been reported. In an adult study from the Mayo Clinic, patients who were treated with medical therapy alone or medical therapy and liver transplant had improved survival. 7 PoPH patients that improve their hemodynamics with medical therapy can successfully undergo liver transplant and may be able to be weaned off pulmonary vasodilator therapy. Unfortunately, three of our patients died before liver transplantation could be considered as an option.
No study has shown superiority of one treatment modality. Treatment itself has consequences that may affect patient outcome. Treatment with continuous intravenous epoprostenol has been reported to be associated with hypersplenism.25,26 Speculation is that the physiologic increase in splanchnic blood flow known to occur in portal hypertension (secondary to an increase in both local and systemic vasodilators) 15 is perhaps potentiated by the use of prostacyclin therapies. Hypersplensim may complicate therapy in PoPH limiting the use of targeted PH therapies in subsets of patients and altering outcomes. As seen in our patient undergoing splenic embolism, huge fluid shifts with splenectomy can lead to high output failure postoperatively, necessitating meticulous attention to fluid and vasodilator management during the perioperative and postoperative periods.
Given the increased risk, subtle presentation, late diagnosis, and poor outcomes of PoPH, multiple adult studies have evaluated the utility of screening all liver cirrhosis patients with echocardiograms to evaluate for evidence of PH.4,12,27 Though echocardiogram is a useful screening tool for at-risk populations, it is insufficient to determine candidacy for liver transplant in affected individuals. Patients identified during echocardiogram screening should undergo cardiac catheterization and appropriate therapy (including targeted medical therapy and closure of intrahepatic shunts) before consideration of liver transplantation.
Secondary to the above experience, our center has developed a collaborative screening protocol between hepatology and PH specialists to periodically evaluate all pediatric patients with portal hypertension aged over ten years, for presence of PH in the hope that early initiation of therapy could alter the outcomes of this rare but life-threatening disorder.
Conclusions
PoPH is a disorder with significant mortality in childhood. Based upon our limited experience, disease progression can be rapid and often presents late. Clinical symptoms are subtle and the majority of patients have severe disease at diagnosis with variable response to treatment. Therapy can be challenging with the development of hypersplenism reported with the use of epoprostenol; however, improvement in PH has also been documented. Multicenter studies of pediatric patients with portal hypertension are required to understand the true extent of this condition and to design appropriate diagnostic protocols and treatment modalities.
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.