
Abstract
Dasatinib is a small-molecule tyrosine kinase inhibitor used in the treatment of hematological malignancies. Pulmonary arterial hypertension (PAH) is a rare but known complication. The mainstay of treatment is cessation of Dasatinib, and while clinical improvement is rapid, complete hemodynamic resolution of pulmonary hypertension (PH) still remains exceedingly uncommon. We present a case of Dasatinib-induced PAH in a woman with chronic myeloid leukemia, who demonstrated rapid and complete clinical and hemodynamic resolution following treatment with combination pulmonary vasodilator therapy using an endothelin receptor antagonist and a phosphodiesterase-5 inhibitor. This case suggests there may be an association between the use of targeted PH medication in combination and the complete resolution of dasatinib-associated PAH, but further investigation is required.
Case description
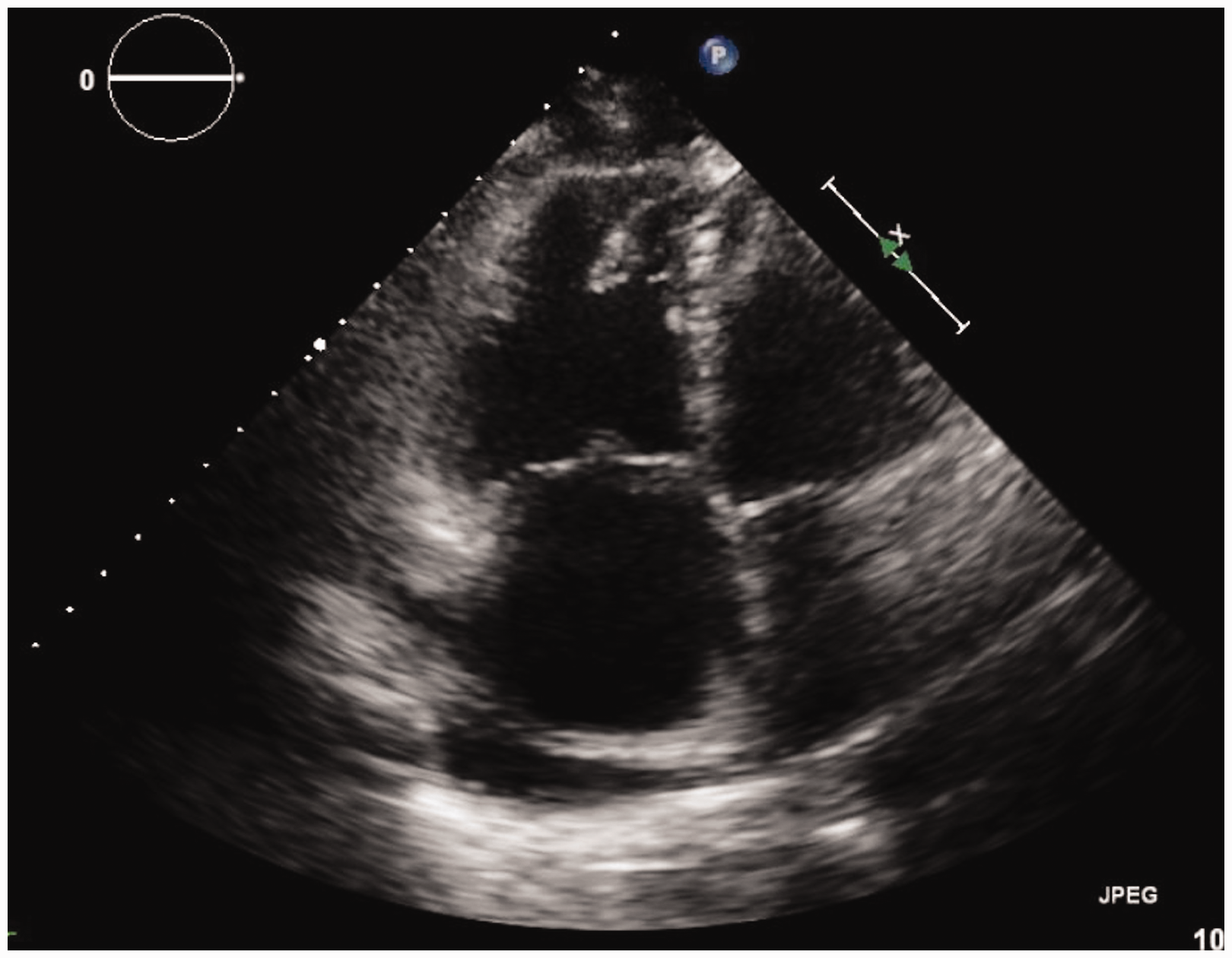
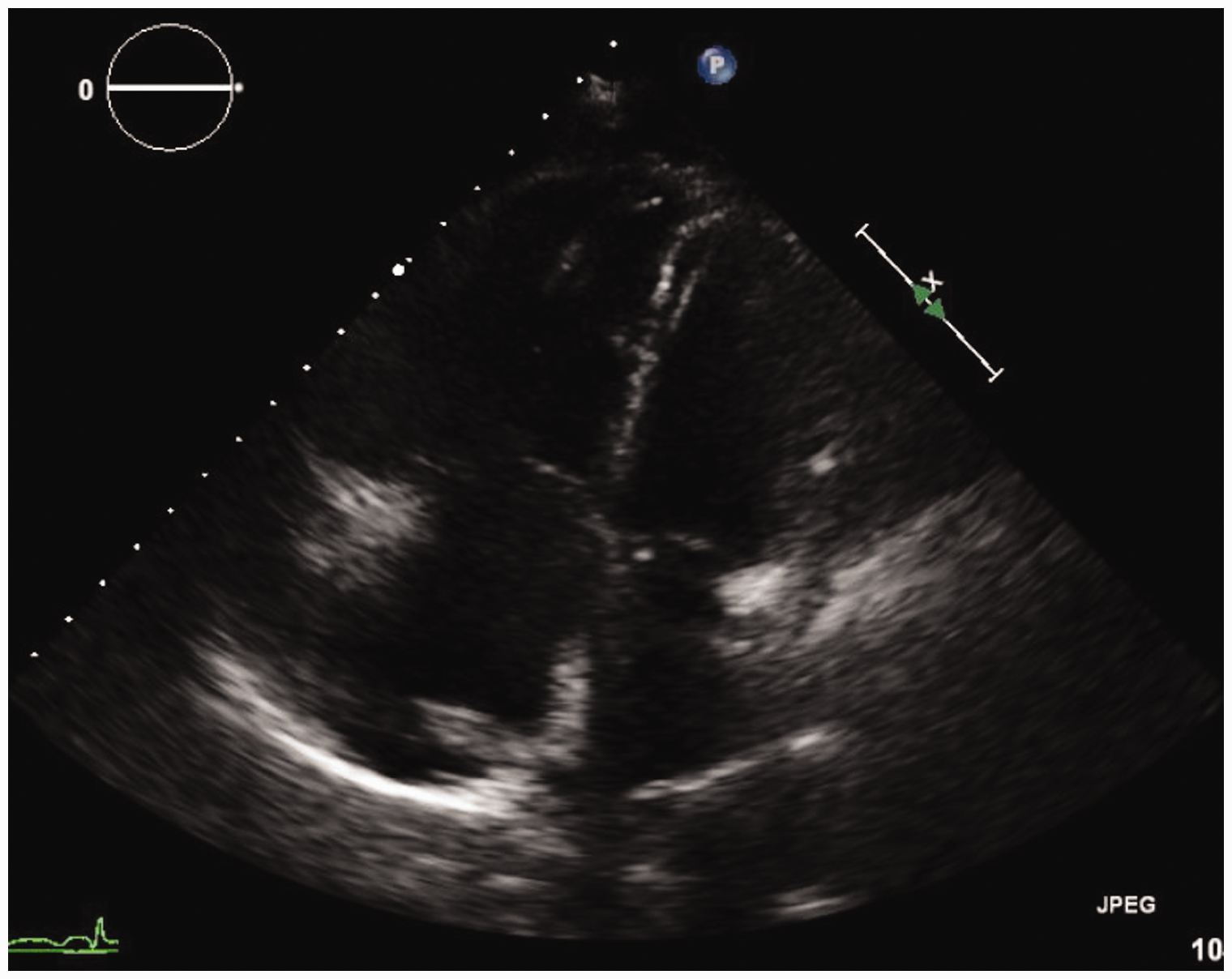
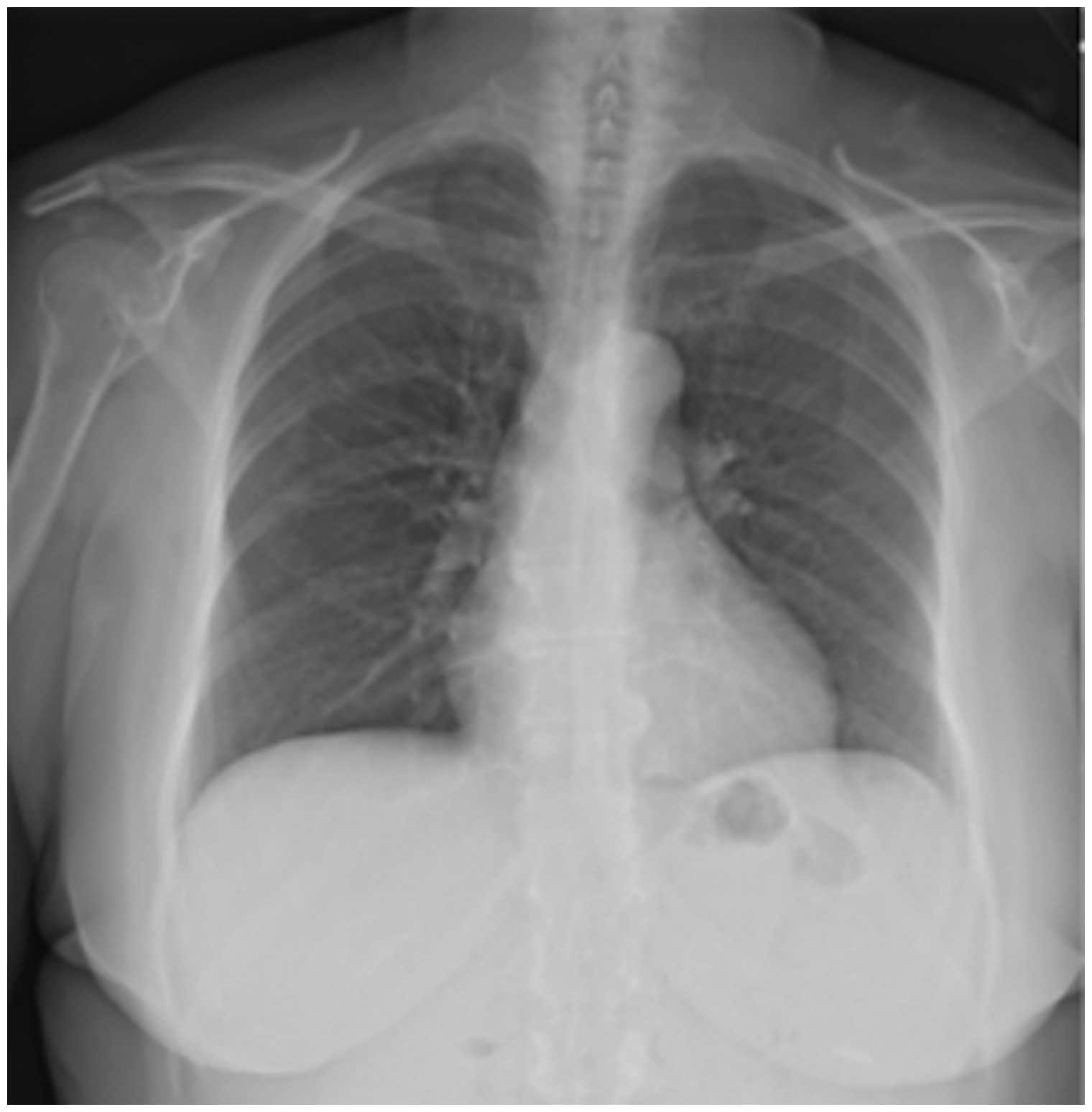
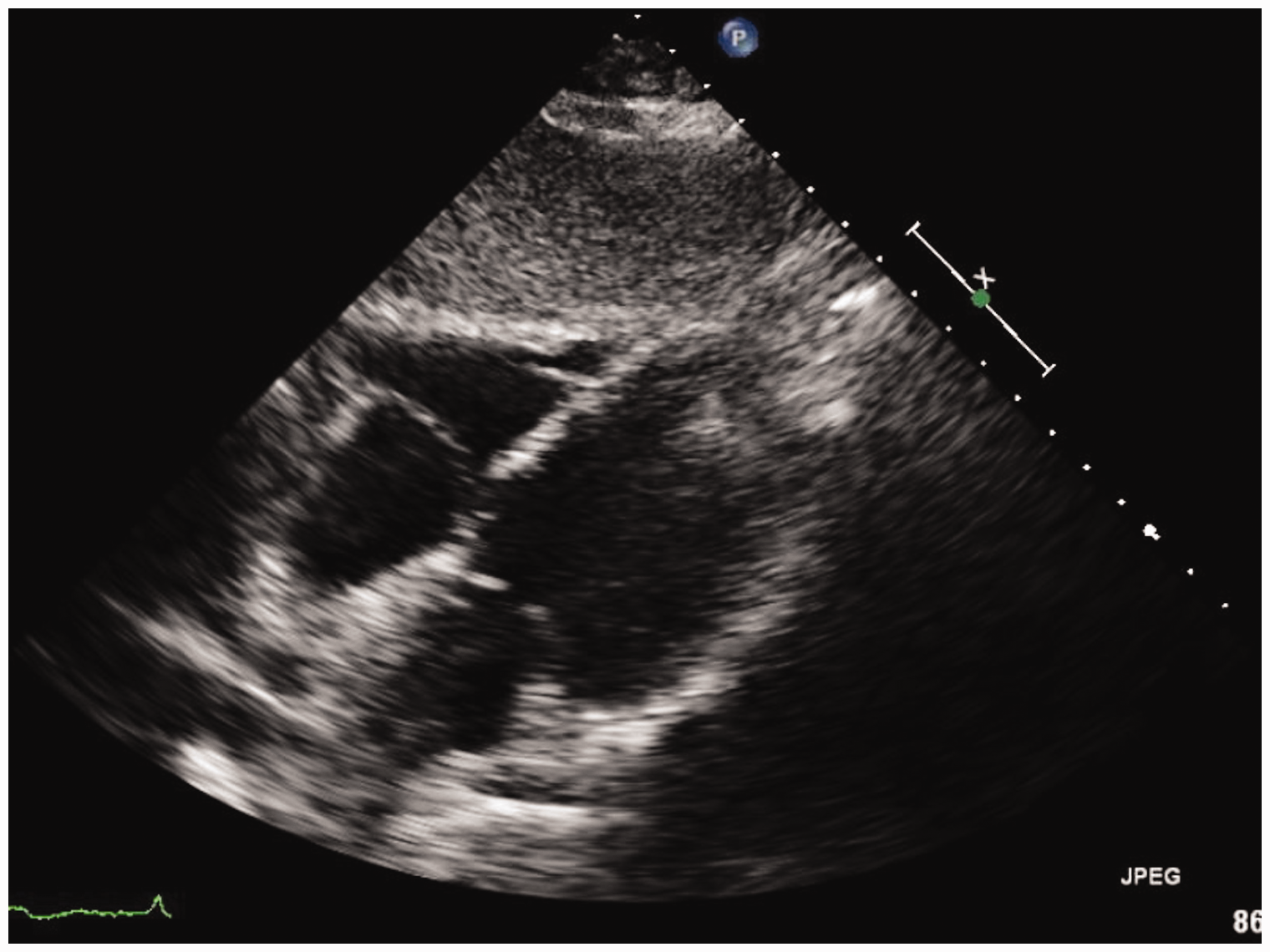
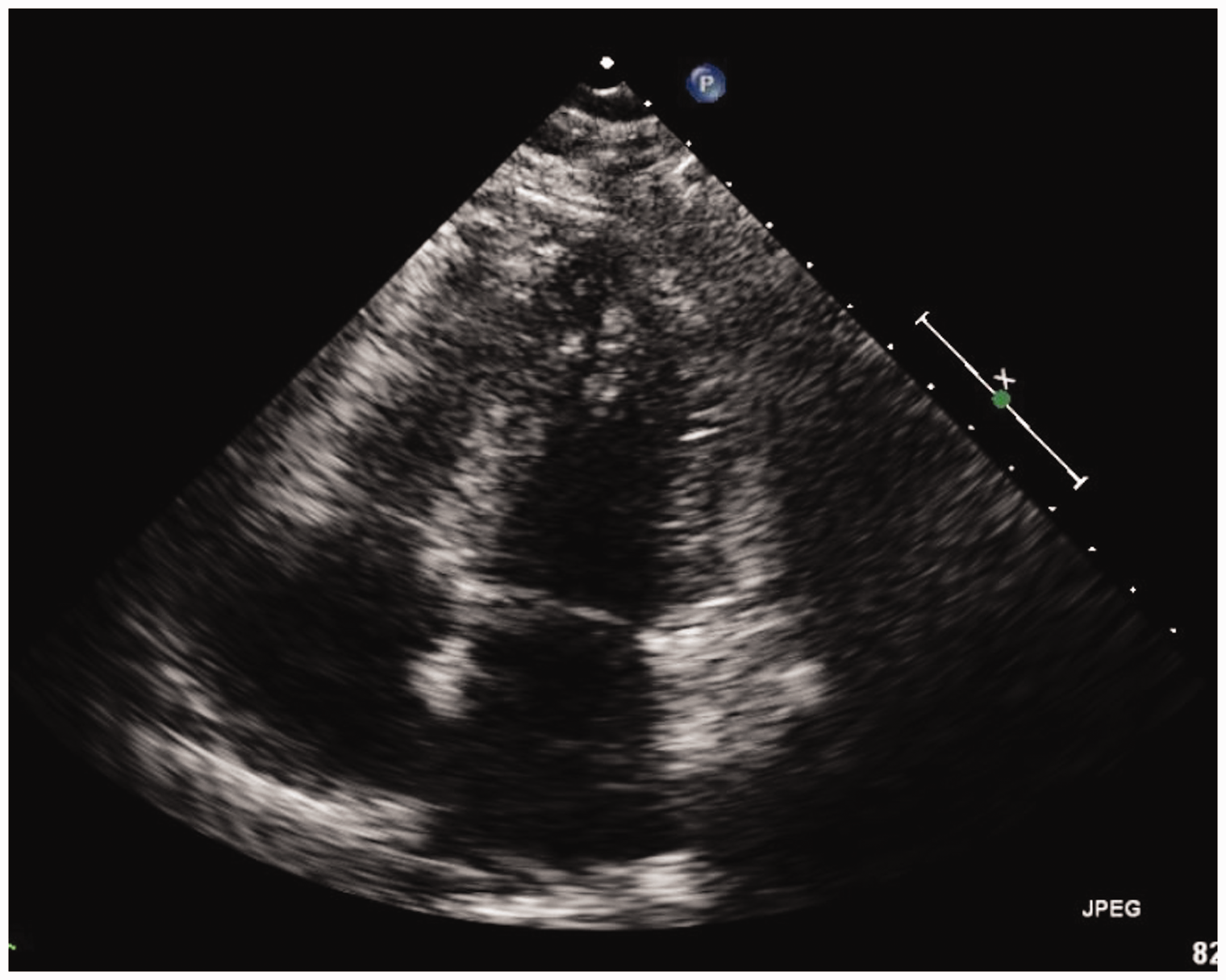
A 61-year-old woman with no past history of cardiopulmonary disease was diagnosed with chronic myeloid leukemia (CML) and initially started on Imatinib therapy. Four years later, a bone marrow biopsy showed progression of CML to precursor B-cell acute lymphoblastic leukemia (ALL) and the patient was switched to therapy with Dasatinib at a dose of 140 mg daily due to disease progression on Imatinib. At this time, the patient also received two cycles of chemotherapy with Cyclophosphamide, Vincristine, Adriamycin, and Dexamethasone. Following this, a repeat bone marrow aspirate showed complete hematologic, cytogenetic, and molecular remission and the patient was continued on Dasatinib therapy. However, 26 months later, the patient developed a persistent dry cough and severe dyspnea on exertion. Initial laboratory testing showed a white blood cell count of 3700, with 46% neutrophils and 45% lymphocytes. Blood cultures were negative for infection, and the patient did not produce adequate sputum for culture. A chest X-ray showed bilateral pleural effusions with cardiomegaly, suggestive of right ventricular failure (Fig. 1). A CT scan was performed, which noted an enlarged central pulmonary artery and bilateral pleural effusions; no other abnormalities concerning the lung parenchyma, pulmonary veins, or interstitium were reported. No pulmonary embolism was seen. An echocardiogram confirmed the presence of a dilated and hypokinetic right ventricle and a dilated right atrium (Figs 2 and 3). Other potential causes of pulmonary hypertension (PH) were excluded by a negative V/Q scan and a review of past records showing normal echocardiographic testing one year prior. A right heart catheterization (RHC) was performed with a mean pulmonary artery pressure (mPAP) of 54 mmHg, pulmonary capillary wedge pressure (PCWP) of 11 mmHg, and pulmonary vascular resistance (PVR) of 18.22 Woods Units, yielding a diagnosis of pulmonary arterial hypertension (PAH). The right atrial (RA) pressure was 19 mmHg, and the cardiac output (CO) by the Fick method was 2.36 L/min (Cardiac index of 1.46 L/min/m2). Vasoreactivity testing was not performed. In clinic follow-up one month after diagnosis, a D-dimer test was done and found to be elevated at 1.73 mg/L (reference = 0–0.5 mg/L); however, a polymerase chain reaction (PCR) test for the BCR-ABL gene was < 0.001% at that time, consistent with her levels while in remission. A complete autoantibody screen was also done and found to be negative (antinuclear antibody, anti-double strand DNA, anti-Smith, ANCA, ribonucleoprotein, anti-histone, SCL-70, chromatin, centromere, SSA/SSB). Her BNP was noted to be 560 pg/mL (reference = 0–100 pg/mL), her 6-minute walk test (6MWT) was 250 m using 2 L oxygen via nasal cannula with a nadir pulse oximetry saturation of 96%, and her functional class was WHO-FC III. Dasatinib was suspected to be the precipitating factor for the patient’s new PAH given the temporal association with the patient’s symptoms and exclusion of other etiologies, and was promptly replaced with Nilotinib (a related second-generation tyrosine kinase inhibitor with a lower rate of drug-induced PAH compared with Dasatinib) to continue therapy for CML. Given the severity of the patient’s symptoms and RHC data, the patient was also started on combination therapy with a phosphodiesterase-5 inhibitor, Tadalafil, at 20 mg daily and an endothelin receptor antagonist, Ambrisentan, at 5 mg daily. The patient was also started on furosemide at 10 mg daily and was not started on systemic anticoagulation. The Tadalafil was up-titrated over a period of four weeks to 40 mg daily, followed by an up-titration of Ambrisentan to 10 mg daily over the following four weeks. The patient experienced marked symptomatic improvement, and repeat chest X-ray (Fig. 4) and echocardiogram four months after beginning therapy showed complete resolution of the PH with a normal right ventricular size and function (Figs 5 and 6). A repeat RHC performed at the same time showed full resolution of PAH, with mPAP of 18 mmHg, PCWP of 9 mmHg, and PVR of 1.96 Woods Units. The RA pressure was 5 mmHg and the CO by the Fick method was 4.59 L/min (cardiac index of 2.51 L/min/m2). Her BNP was measured in clinic at this time and had decreased to 18 pg/mL, her 6MWT had improved to 425 m on room air with a nadir pulse oximetry saturation of 96%, and her functional class was WHO-FC I. Given the marked improvement both symptomatically and hemodynamically, Ambrisentan and furosemide were discontinued and Tadalafil was weaned sequentially over the following four months. Despite the cessation of her pulmonary vasodilator therapy, the patient did not have a return of her dyspnea or exercise intolerance. She continues to remain symptom-free after five months without targeted PH therapy.
Chest X-ray at time of initial diagnosis of PAH. Echocardiography (apical two-chamber view) at time of initial diagnosis of PAH. Echocardiography (apical four-chamber view) at time of initial diagnosis of PAH. Chest X-ray after cessation of PAH-specific therapy. Echocardiography (apical two-chamber view) after cessation of PAH-specific therapy. Echocardiography (apical four-chamber view) after cessation of PAH-specific therapy.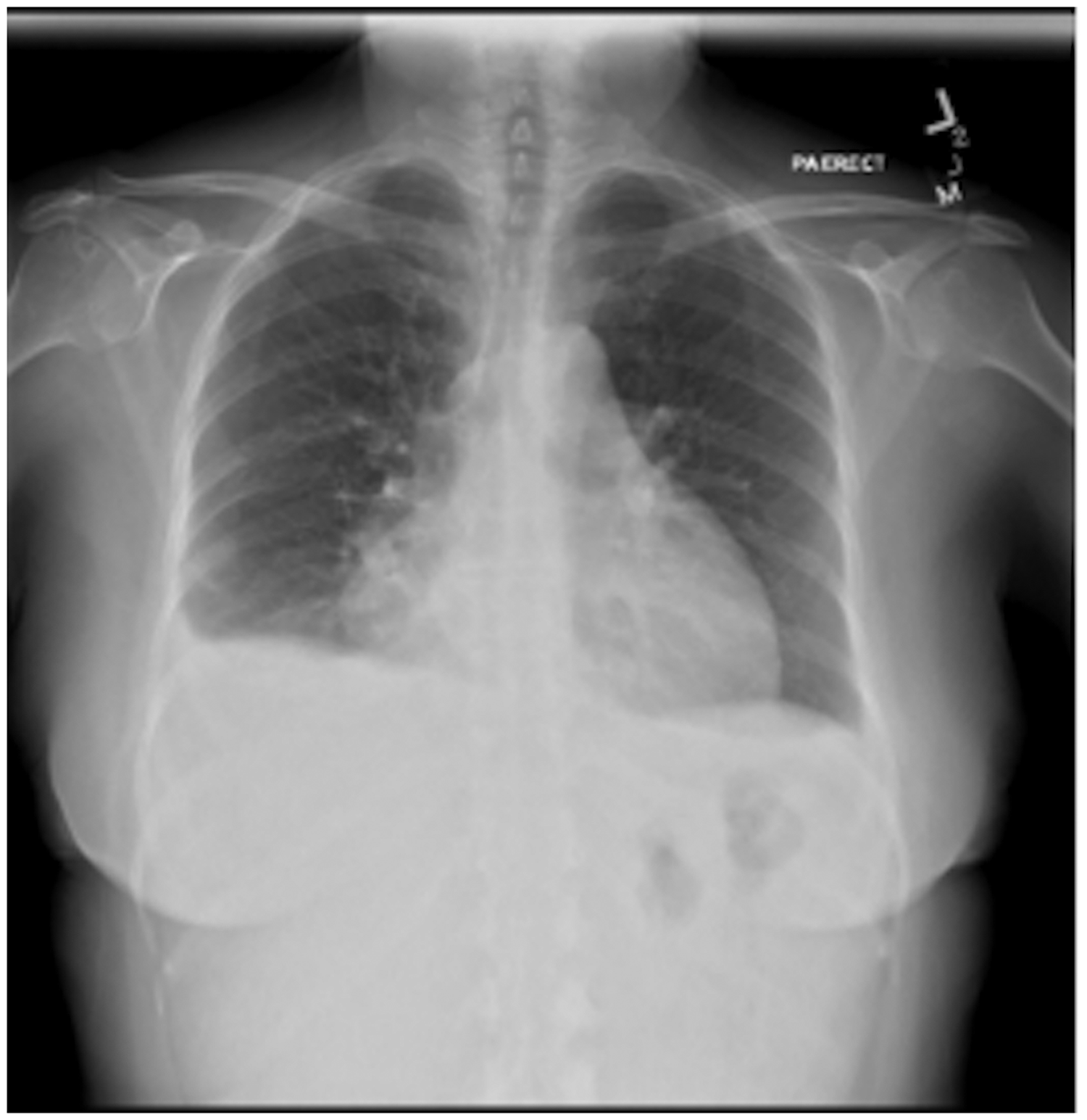
Discussion
Dasatinib is a potent oral small-molecule tyrosine kinase inhibitor of the BCR-ABL oncogene used in the treatment of diseases such as CML and ALL.1,2 PAH has been reported as a rare complication of Dasatinib therapy that can occur at any time during the treatment course, ranging from months to over one year after initiation. 3
Drug-induced PH secondary to Dasatinib is rare, and the exact mechanism is unknown but thought to be related to the platelet-derived growth factor pathway (PDGF) and the Src family of kinases. 4 While its main therapeutic effect in CML patients is high-affinity inhibition of the BCR-Abl oncoprotein, Dasatinib is a non-specific tyrosine kinase inhibitor and also inhibits a number of different kinases, including Src and the receptor for PDGF. 2 In animal models of PH, dysfunction of the PDGF pathway has been involved in the development of PH, and the Src family of kinases is involved in smooth muscle cell proliferation and vasoconstriction of the pulmonary vasculature. More recent work has shown a direct link between Dasatinib exposure and pulmonary endothelial cell damage, dysfunction, and ultimately apoptosis in both a rat PAH model and cultured human pulmonary endothelial cells. Interestingly, treatment with Imatinib, a related tyrosine kinase inhibitor, did not have the same effect. 5 Indeed, evidence suggests related inhibitors such as Nilotinib and Imatinib do not have the same frequency of drug-induced pulmonary vascular dysfunction as Dasatinib, and the pulmonary vascular toxicity for Dasatinib is molecule-specific. 6 Some related tyrosine kinase inhibitors such as Imatinib have also been used as treatment for PH, highlighting how much is still unclear regarding the exact mechanism of drug-induced PH from Dasatinib. 7
While drug-induced PAH secondary to Dasatinib is accompanied by serious clinical complications, evidence suggests it may be reversible to some degree. 8 To date, the cornerstone of treatment is cessation of the offending drug in question; targeted PH medication is frequently used to address the underlying hemodynamic and functional abnormalities present. While this leads to rapid improvement in both symptoms of right heart failure and hemodynamic measures of PH severity, complete resolution of PH is fairly uncommon. In an analysis of a French Registry of nine Dasatinib-induced PAH cases, the initial hemodynamic abnormalities seen on diagnostic RHC (median mPAP of 46 mmHg, median PVR of 5.9 Woods Units) improved significantly with cessation of Dasatinib. However, none of the patients had complete resolution of PH at 15-month follow-up. This was despite two patients receiving treatment with endothelin receptor antagonists and one patient receiving therapy with calcium channel blockers. 9
Similarly, in an analysis of the Bristol-Meyers Squibb database of adverse drug events due to Dasatinib, 41 cases of PAH confirmed by RHC due to Dasatinib were identified, of which there was symptomatic or echocardiographic resolution of PAH in 21 cases. Only five patients had a subsequent RHC following drug cessation, of which none had complete resolution of PAH as defined by mPAP < 25 mmHg at rest. While there was no relationship between the duration of Dasatinib exposure and improvement of PAH (ranging from one week to 75 months of Dasatinib therapy), the majority of improved cases (13 of 21, 62%) involved treatment with pulmonary vasodilator medications such as endothelin receptor antagonists (ERA) or phosphodiesterase-5 inhibitors (PDE-5i). Of note, two of the improved cases used combination targeted PH therapy with a PDE-5i and an ERA simultaneously, showing rapid clinical resolution in less than one month. Unfortunately, following clinical resolution neither case had a subsequent RHC to demonstrate hemodynamic resolution of PAH. 8
A PubMed/Medline search revealed several case reports of Dasatinib-induced PAH, some of which resolved with cessation of the drug alone,10,11 and others that resolved with the addition of PAH-specific medications.12,13 These reports differ in both the duration of Dasatinib treatment prior to the development of PAH, the type of PAH-specific therapy used, as well as the duration of time required for resolution of PAH; however, none of these published cases involved the use of combination therapy. Notably only two cases confirmed complete hemodynamic resolution of PAH with RHC, and both involved therapy with a PDE-5i (Sildenafil). The time to hemodynamic resolution was seven months and six months, respectively.12,13 Currently there are no data on the use of PAH-specific medication to increase the extent and/or rapidity of Dasatinib-induced PAH resolution, either alone or in combination.
As noted above, our patient was not anticoagulated, and currently there is conflicting evidence regarding systemic anticoagulation in PAH. The publication of the Registry to Evaluate Early and Long-term PAH disease Management (REVEAL) showed there was no survival benefit to systemic anticoagulation in idiopathic PAH (IPAH) patients, and a trend towards increased mortality in patients with systemic sclerosis-associated PAH (SSc-PAH). This is contrary to the Comparative, Prospective Registry of Newly Initiated Therapies for Pulmonary Hypertension (COMPERA) registry, which showed a statistically significant survival benefit at three years in IPAH patients on systemic anticoagulation. Given the uncertainty regarding the survival benefit in IPAH patients, as well as the concern for increased mortality in SSc-PAH exposed to systemic anticoagulation, the decision is currently left to the provider and patient on an individualized basis and the optimal strategy in drug-induced PAH patients is unknown.14,15
The use of combination targeted PH therapy in PAH patients is supported by the AMBITION trial, where initial combination therapy resulted in a 50% reduction in the rate of clinical failure when compared with monotherapy in previously untreated patients with PAH. 16 As noted above, complete resolution of Dasatinib-induced PH is exceedingly uncommon, but registries and case reports suggest there is an association between the use of targeted PH medications and complete resolution. We believe this case suggests a potential association between the use of combination targeted PH therapy and the complete resolution of Dasatinib-associated PAH. It is unclear if combination therapy may be useful in increasing the chances of complete resolution after drug cessation, and additional research is warranted to investigate any potential association that may exist.
Therapy with Dasatinib, a small-molecule tyrosine kinase inhibitor used in the treatment of CML and ALL, is known to cause drug-induced PAH. The mainstay of treatment is cessation of Dasatinib, which can lead to improvement in the severity of PAH but does not typically result in complete resolution. As seen in this case, there may be an association between the use of targeted PH medications in combination and the complete resolution of PAH, but further investigation is required.
Footnotes
Conflict of interest
The authors declare that there are no conflicts of interest.
Funding
This research received no specific grant from any funding body in the public, commercial, or not-for-profit sectors.