
Abstract
Within the cohort of patients suffering from idiopathic pulmonary arterial hypertension (IPAH) is a group that responds dramatically (VR-PAH) to an acute vasodilator challenge and that has excellent long-term hemodynamic improvement and prognosis on high dose calcium channel blockers compared with vasodilator non-responders (VN-PAH). For the purposes of diagnosing VR-PAH, there is to date no test to replace the acute vasodilator challenge. However, recent studies have identified markers that may aid in the identification of VR-PAH, including peripheral blood lymphocyte RNA expression levels of desmogelin-2 and Ras homolog gene family member Q, and plasma levels of provirus integration site for Moloney murine leukemia virus. Genome wide-array studies of peripheral blood DNA have demonstrated differences in disease specific genetic variants between VR-PAH and NR-PAH, with particular convergence on cytoskeletal function pathways and Wnt signaling pathways. These studies offer hope for future non-invasive identification of VR-PAH, and insights into pathogenesis that may lead to novel therapies. Examination of the degree of pulmonary microvascular perfusion in PAH has offered additional insights. During the acute vasodilator challenge, VR-PAH patients demonstrate true vasodilation with recruitment and increased perfusion of the capillary bed, while VN-PAH patients are unable to recruit vasculature. In the very few reports of lung histology, VR-PAH has more medial thickening in the precapillary arterioles, while VN-PAH has the classic histology of PAH, including intimal thickening. VR-PAH is a disorder with a phenotype distinct from VN-PAH and other types of PAH, and should be considered separately in the classification of PAH.
Keywords
Introduction
From many aspects, idiopathic PAH (IPAH) remains an enigma. In most patients, pulmonary microvascular cell proliferation and abnormalities of endothelial mediator production result in precapillary vascular luminal restriction, increased pulmonary vascular resistance (PVR), and a loss of perfused downstream pulmonary capillary surface area.1,2 The underlying causes and pathogenic mechanisms are not fully elucidated. The diagnosis of IPAH is based on having a mean pulmonary arterial pressure (mPAP) ≥ 25 mm Hg, pulmonary artery wedge pressure (PAWP) ≤ 15 mmHg, and PVR > 3 Wood units, after exclusion of other causes of pulmonary hypertension. 3 Recent identification of associated gene mutations allows for very specific diagnosis in some patients. 4
Within the cohort of IPAH patients is a group of otherwise clinically indistinguishable subjects that behave differently during an acute vasodilator challenge. Unlike the rest, this minority of IPAH patients demonstrates a marked vasodilator response, has a sustained response to high-dose oral calcium blocker therapy, and has a seemingly superb long-term outcome and survival.5–7 Recent physiologic and molecular studies suggest that this group with vasodilator-responsive PAH (VR-PAH) represents a distinct phenotype within the IPAH population.8–10
History
The description of primary pulmonary hypertension (PPH, now termed IPAH) and its hemodynamic analysis led to the observation that some patients responded acutely to pulmonary vasodilators.11–13 Many systemic antihypertensive agents were subsequently tested during acute vasodilator challenges and as chronic therapy, including hydralazine, phentolamine, captopril, diazoxide, isoproterenol, conventional-dose nifedipine, tolazoline, and others.14–26 They were abandoned when it became evident that they generally had little long-term beneficial effect on pulmonary hemodynamics in IPAH patients but had adverse effects including lowering systemic blood pressure, sometimes to dangerous levels.27–36 Moreover, they did not provide criteria for predicting a chronic vasoresponder group.
37
Although the possibility of a response was not linked to the severity of PVR elevation, there was a suggestion that participants with a less abnormal resting PVR had a greater likelihood of responding
37
(Fig. 1).
Frequency distribution of patients with primary pulmonary hypertension (PPH, now termed IPAH) in a review of prior publications, ranked by PVR (Wood units). The dark areas represent the number of patients that demonstrated some degree of vasoresponsiveness during an acute vasodilator challenge. Reproduced with permission of the American Thoracic Society, from Reeves et al.
37
Copyright © 2017 American Thoracic Society. The American Review of Respiratory Disease is an official journal of the American Thoracic Society.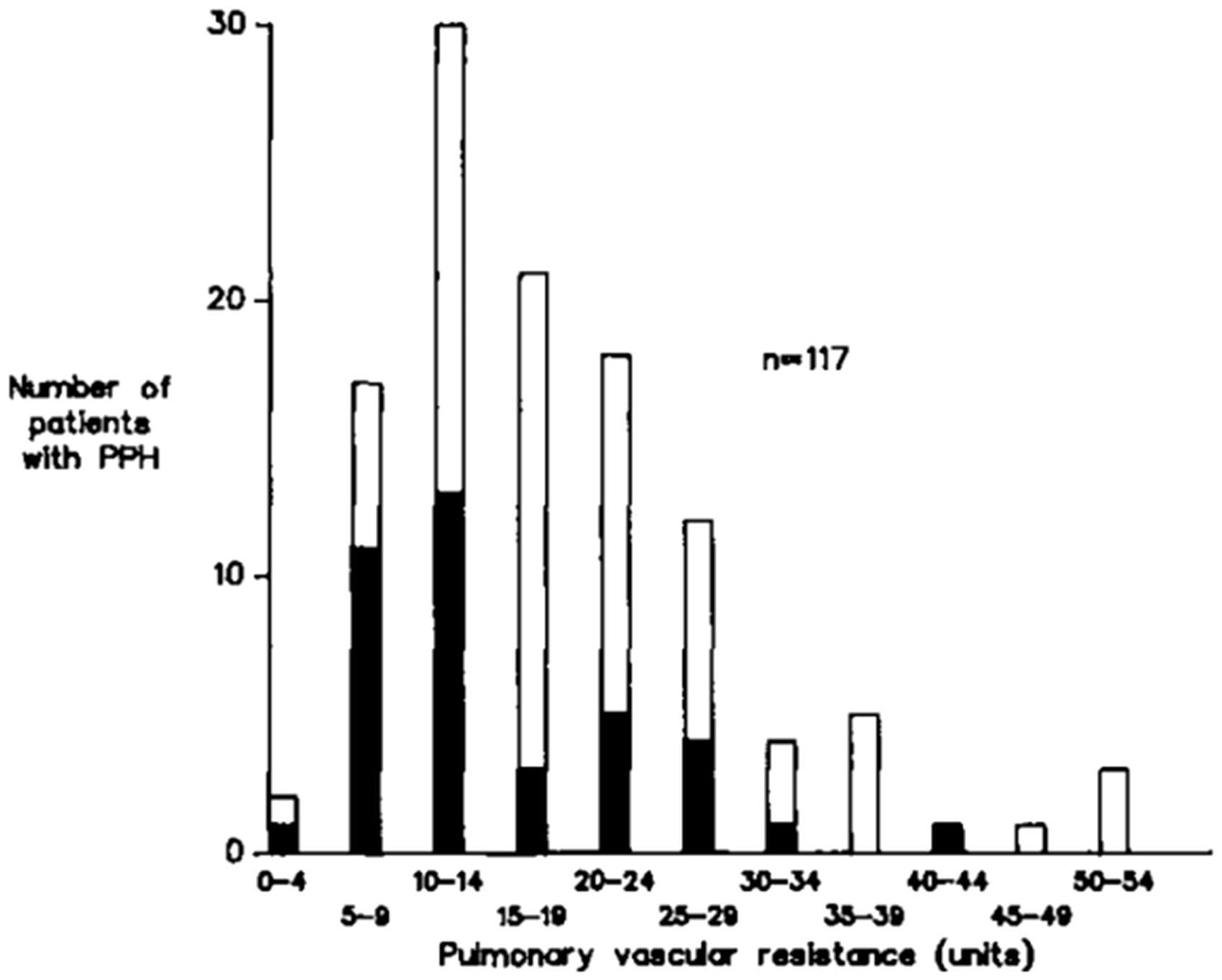
There are several possibilities for these failures. First, a given medication may simply have been ineffective. Second, the medication might have been somewhat effective, but only in a minority of patients, and the evidence of benefit was obscured within a group that contained a large number of vasodilator non-responsive (VN-PAH) patients. Third, the criteria for response may have been incorrect. Many of the studies were able to demonstrate an increase in cardiac output with at times very significant reductions in calculated PVR, but without any decrease in pulmonary artery pressure.15,18 This was termed a “resistance” response, as opposed to a “pressure” response which consists of a true fall in PAP with either an increase or no change in cardiac output, also resulting in a decrease in calculated PVR. As discussed below, these two patterns of response have very different physiologic implications in terms of distention and recruitment of pulmonary microvasculature. A careful analysis of multiple studies suggested that using a “resistance” response as an indicator of therapeutic benefit was insufficient and misleading and, by current criteria, the participants would be considered as VN-PAH patients. 38
The need for safer and effective agents for acute vasodilator testing led to the study of prostanoids, inhaled nitric oxide, and adenosine.39–49 All have a short duration of action, none impair myocardial contractility, and inhaled nitric oxide has no systemic vasodilator effects. Adenosine is given intravenously, while the prostanoids may be given intravenously or via inhalation. While not identical, the vasodilator response to one type of agent correlates with the others.39,40,42,47 Guidelines for use of these agents as acute vasodilators have recently been updated. 50 Inhaled nitric oxide at 20 parts per million (ppm) is the preferred testing agent, although some studies have used increments up to 80 ppm. Intravenous epoprostenol is started at 2 ng/kg/min, with increments of 2 ng/kg/min every 10 min up to a maximum of 16 ng/kg/min, watching for effectiveness, or side effects including systemic hypotension. Adenosine infusions are started at 0.001 mg/kg/min, with increments of 0.01–0.02 mg/kg/min every 10 min, up to a maximum dose of 0.05 mg/kg/min.
In 1987, Rich and Brundage described 13 patients with PPH that were challenged with hourly oral doses of the calcium channel blockers nifedipine or diltiazem until they experienced adverse effects or until they demonstrated a positive response defined as a minimum 50% reduction in PVR and a minimum 33% decrease in mPAP.
5
The participants had essentially no response to a single conventional dose of the calcium blockers. However, after hourly dosing, eight patients (62%) met the response criteria defined above. Five patients had hemodynamic follow-up at one year, with sustained benefit in four of them, on daily doses of calcium channel blockers as high as 180 mg of nifedipine or 720 mg of diltiazem. Another responder died after reducing her nifedipine dose to 40 mg/day. In a subsequent study of 64 patients, using a criterion of a minimum 20% decrease in both PAP and PVR, 17 (27%) were responders.
6
Thirteen of the 17 had sustained hemodynamic benefit in long-term follow-up, in some cases up to five years (Fig. 2). The survival of the responders was 94% at five years compared with a predicted three-year survival of 55%. Another report of 47 patients using the same criteria described a response rate of 32%.
51
The importance of a “pressure” response versus a “resistance” response was emphasized.
Effects of high-dose calcium blocker therapy in a group of VR-PAH patients, showing effects up to a five-year period in: pulmonary artery pressure (left); PVR (Wood units, center), and survival (right). Reproduced with permission from Massachusetts Medical Society, from Rich et al.
6
Copyright © 2017 Massachusetts Medical Society.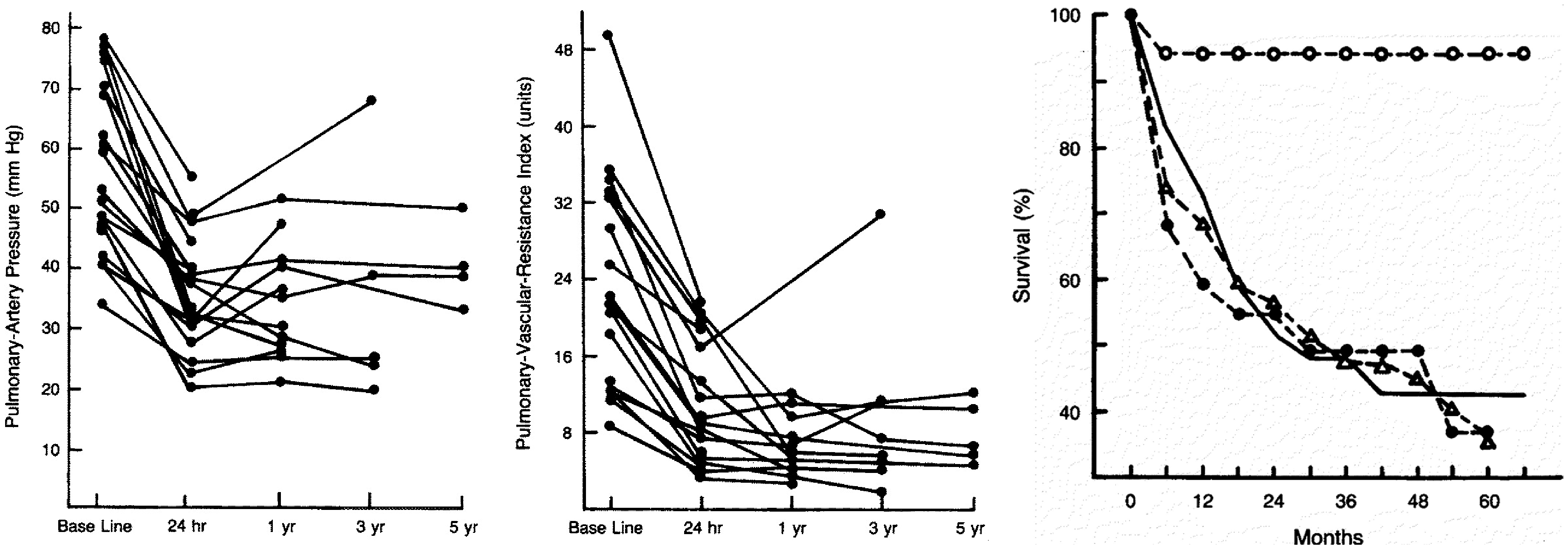
In 2005, Sitbon et al. described 557 IPAH patients that had undergone vasodilator testing with intravenous epoprostenol or inhaled nitric oxide.
7
They employed the traditional criteria of a minimum 20% decrease in mPAP and in PVR. They defined a long-term calcium channel blocker response as the patients being in World Health Organization (WHO) functional class I or II at one year. Seventy patients (12.5%) met the criteria for acute vasoresponsiveness. However, only 38 (6.8%) had long-term improvement on calcium channel blockers. In those patients, the average seven-year survival was 97%, and they had less severe hemodynamic abnormalities at baseline than the patients who failed calcium blockers. The study led to a reconsideration of the definition of a vasodilator responder. Based on the data, an acute response is now defined as a decrease in mPAP ≥ 10 mmHg to a mPAP of ≤ 40 mmHg, with an unchanged or increased cardiac output.
50
PVR was not a predictor of long-term response. Another study explored the relationship between acute vasodilator responsiveness and long-term calcium blocker responsiveness in patients with PAH other than idiopathic.
52
With the exception of PAH related to anorexigens, the incidence of acute responsiveness was either low or did not predict a long-term response, and the incidence of long-term responses was very low (Fig. 3). The current guidelines suggest acute vasodilator testing and long-term use of calcium channel blockers only in patients with IPAH, heritable PAH, and drug-induced PAH.
50
Percentage of PAH patients responding to an acute vasodilator challenge and to long-term calcium channel blocker (CCB) therapy, ranked by the condition associated with the PAH (on the vertical axis). Only PAH associated with appetite suppressants has a long-term response to CCB. Reproduced with permission from the European Society of Cardiology and Oxford University Press, from Montani et al.
52
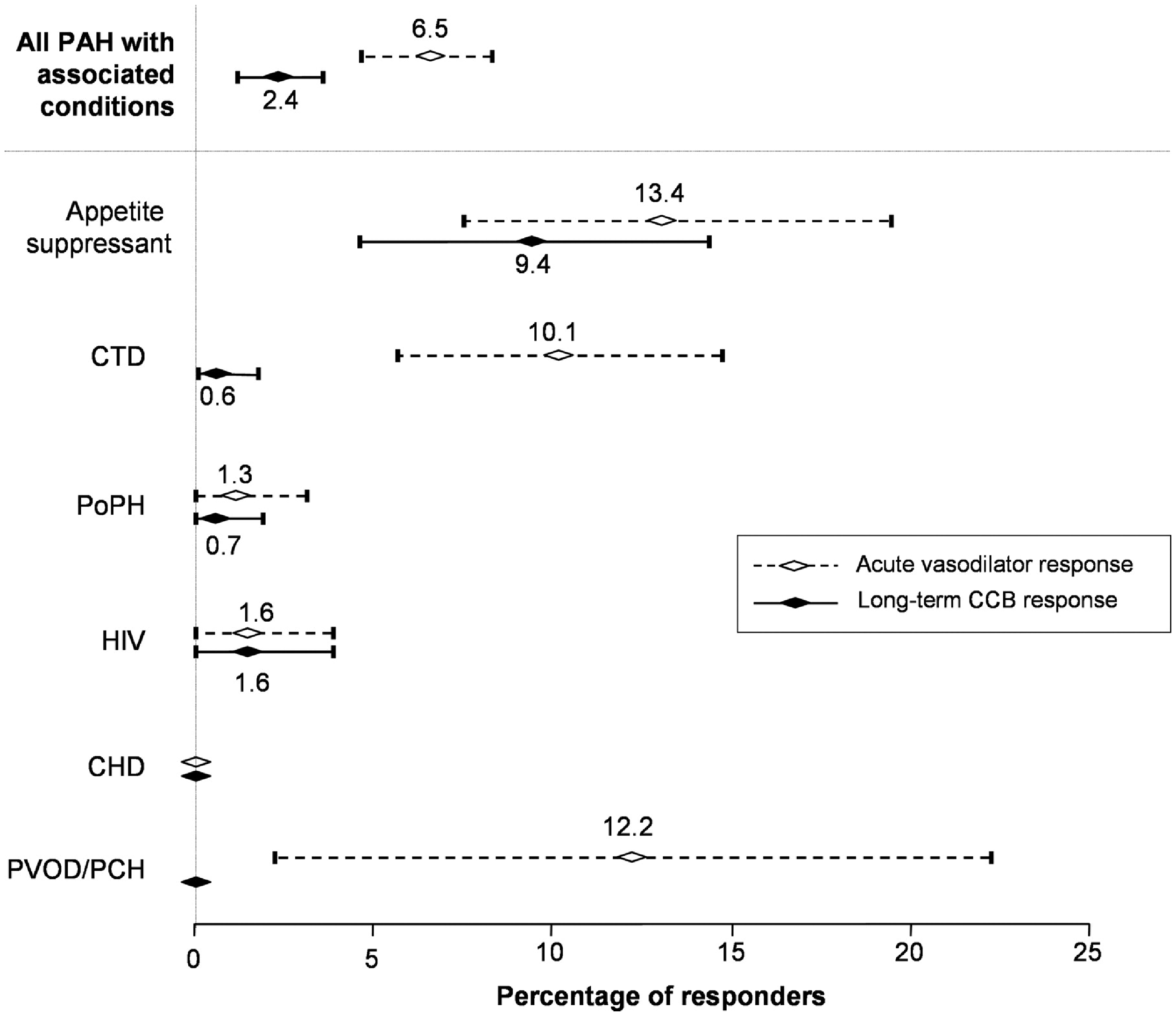
Calcium channel blockers should not be used as acute vasodilator testing agents as they may have acute systemic vasodilator and negative inotropic effects, resulting in decreased cardiac output or hypotension, and they have a long duration of action should deleterious effects occur.
Blood and genetic markers, and histology, of vasoreactivity and long-term response
Other than with the acute vasodilator challenge, which is invasive and costly, there are currently no simple methods that can identify VR-PAH patients. However, recent studies are beginning to provide a molecular basis for the distinct behavior of VR-PAH patients and offer the hope for a simple screening tool and for understanding the cause(s) of VR-PAH. Hemnes et al. studied cultured peripheral blood lymphocytes from IPAH patients and, based on gene expression RNA patterns, formed a decision tree that correctly identified VR-PAH patients.
9
The trees were then validated using an IPAH cohort from a second PH specialty center (Fig. 4). While there were significant differences in expression in 13 genes, the expression levels of two genes proved central and sufficient to the development of the decision tree: DSG2 and RHOQ. Desmoglein-2 (DSG2) is a desmosomal cadherin that affects Wnt/β-catenin signaling, cell–cell adhesion, and calcium binding. Abnormal Wnt/β-catenin signaling may contribute to the pathogenesis of IPAH.53–55 Mutations in DSG2 have been described in arrhythmogenic right ventricular cardiomyopathy (ARVC).56–59 Interestingly, a mutation in a gene coding for phospholamban has also been described in ARVC.
60
Phospholamban regulates the sarcoplasmic reticulum calcium load and it has been proposed that this may result in maladaptive remodeling of the intercalated disk. This finding of altered cellular calcium handling may be relevant to VR-PAH, given the necessary unconventionally high calcium channel blocker doses required to maintain therapeutic benefit. Ras homolog gene family, member Q (RHOQ), is a cytoskeletal GTPase involved in protein transport and insulin-mediated signaling.61,62 Abnormalities in glucose handling have been described in PAH.63,64 Taken together, when expression levels relative to hypoxanthine guanine phosphoribosyl tranferase expression were measured by PCR, a RHOQ level ≥ 2.8 and a DSG2 level ≥ −7.8 as primary genes, with TPD52 ≥ −0.39 as a secondary gene, correctly identified VR-PAH, with no false positives or negatives. This technique represents a significant advance in detecting VR-PAH using a peripheral blood sample.
An example of a screening strategy to distinguish VR-PAH from NR-PAH, based on gene expression patterns in RNA from peripheral blood lymphocytes. Measurements of relative expression of desmoglein-2 (DSG2) and Ras homolog gene family member Q (RHOQ), compared with expression of hypoxanthine guanine phosphoribosyl transferase, correctly identified VR-PAH, with no false positives or negatives. Reproduced with permission from Hemnes et al.
9
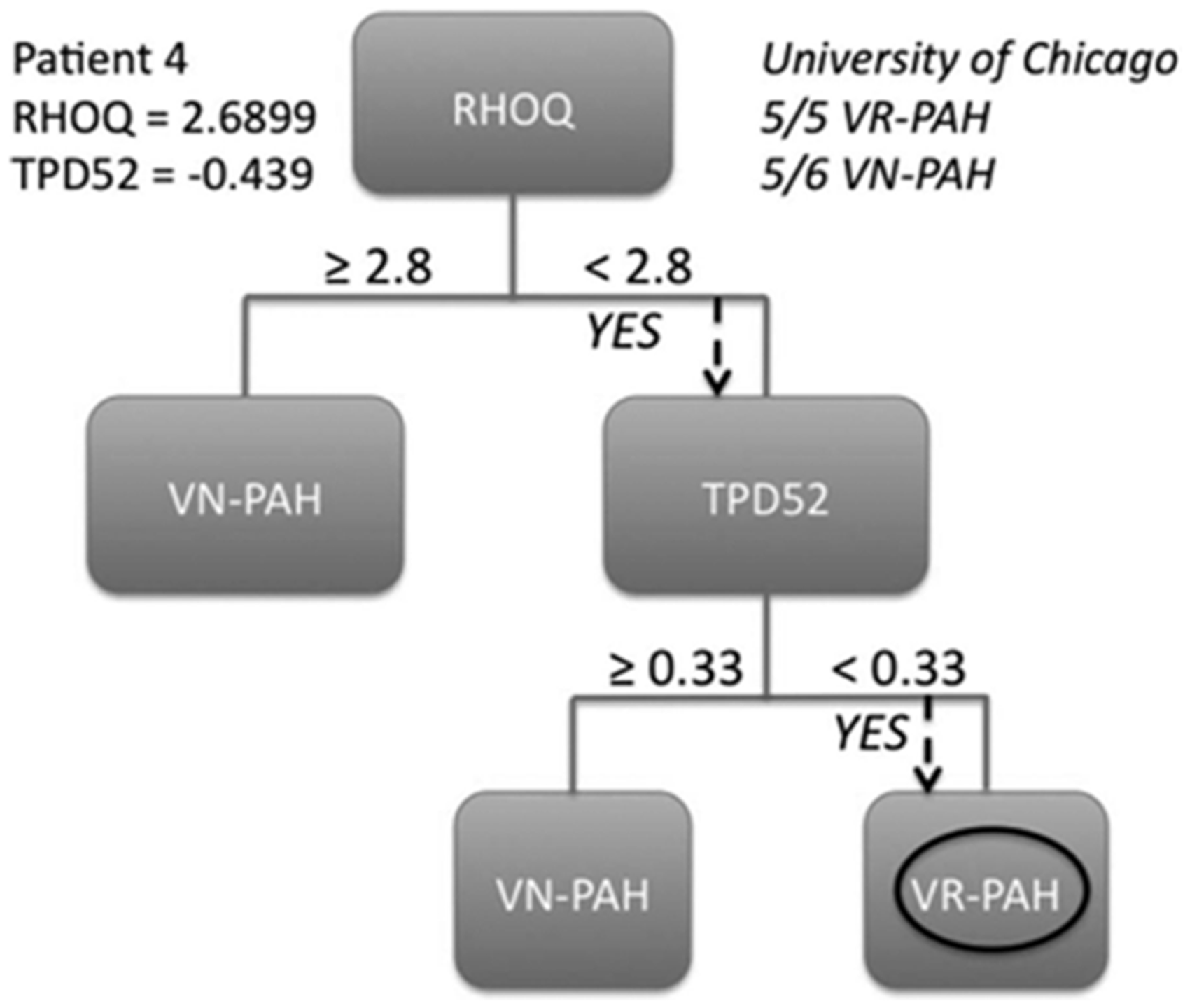
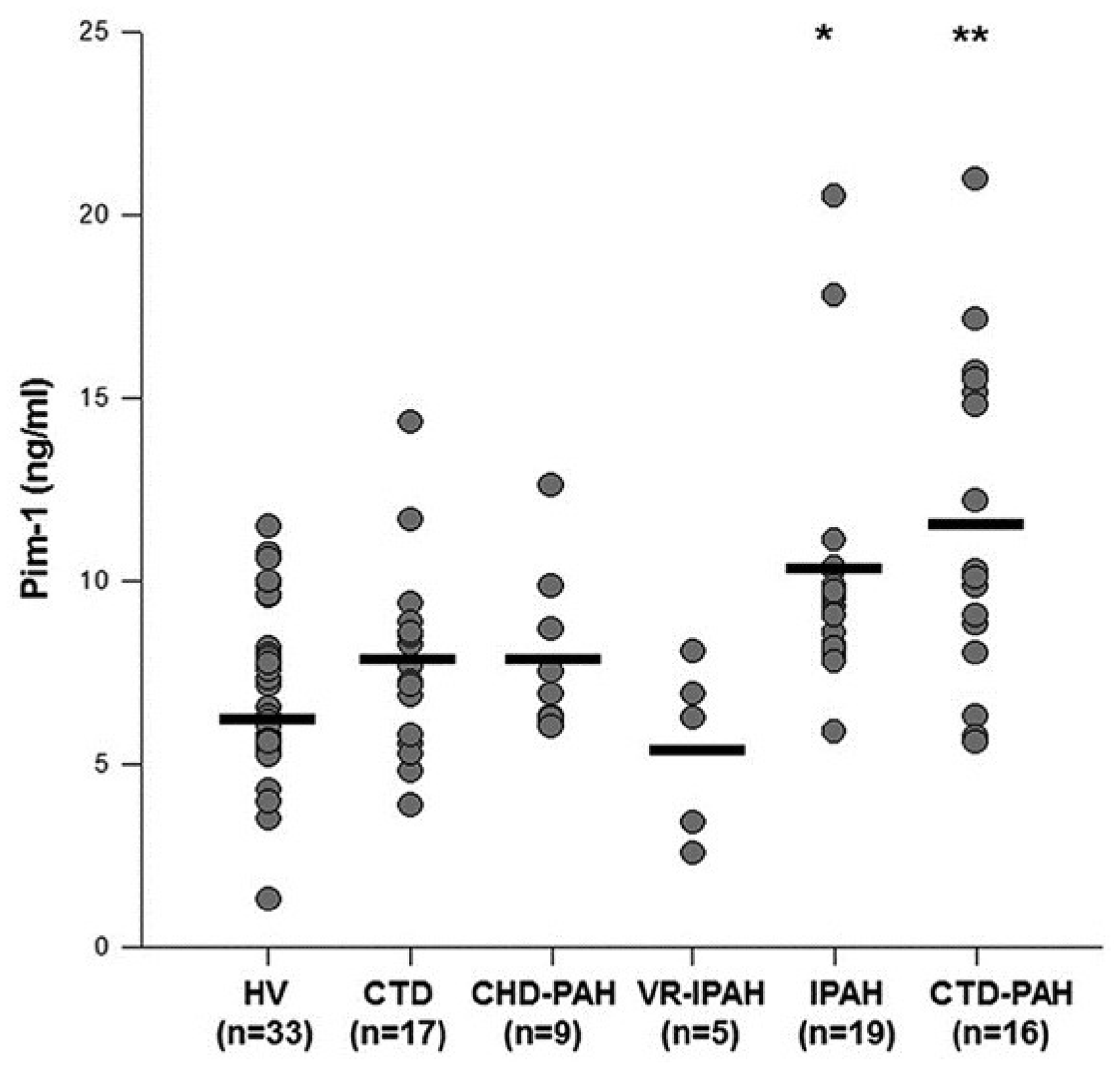
In another approach, Renard et al. demonstrated that plasma levels of provirus integration site for Moloney murine leukemia virus (Pim-1) could differentiate VR-PAH from NR-PAH65 (Fig. 5). Pim-1 is a proto-oncogene that activates the NFAT/STAT3 pathway and may contribute to the pathogenesis of PAH, at least in its proliferative form.
66
Participants with VR-PAH had levels of Pim-1 similar to those of healthy volunteers. By contrast, patients with NR-PAH and PAH from connective tissue disease had higher levels of Pim-1. The predictive threshold for distinguishing NR-PAH from VR-PAH was >8.2 ng/mL, with a sensitivity of 79% and a specificity of 100%. These results are intriguing because they may allow for separation of VR-PAH from VN-PAH via a peripheral blood test. It is unknown whether combining the above approaches of Hemnes and Renard would provide even greater discriminatory power.
Plasma levels of provirus integration site for Moloney murine leukemia virus (Pim-1) in healthy volunteers (HV), connective tissue disease (CTD) without or with PAH, PAH associated with congenital heart disease (CHD-PAH), and idiopathic PAH which was vasoresponsive (VR-IPAH) or non-responsive (NR-PAH). Pim-1 levels in VR-IPAH are similar to those of healthy volunteers. Reproduced with permission from Renard et al.
65
Examination of genetic variants may also help. Using peripheral blood-derived DNA, Hemnes et al. analyzed IPAH patients (17 VR-PAH, 19 VN-PAH) and identified 1580 genetic variants that were specific to the disease.
10
With whole exome sequencing, they found that the variants converged on cytoskeletal function pathways and the Wnt signaling pathway (Fig. 6). Compared with VN-PAH, vascular smooth muscle contraction-related genes were enriched in VR-PAH. The authors suggest that, if there is a shared gene regulating smooth muscle contraction, this may explain why vasodilators with different mechanisms of action (i.e. prostanoids, nitric oxide, adenosine, and calcium blockers) are effective in VR-PAH. Moreover, since the number of variants was higher in VR-PAH than VN-PAH, VR-PAH might have a completely genetic disposition or VR-PAH might result from pathways related to VN-PAH being further modified. It is currently unknown which possibility is correct. All the above studies identify markers for VR-PAH versus NR-PAH, but they also offer mechanistic insights that may ultimately lead to better therapies.
Examination of genetic variants in peripheral blood-derived DNA from VR-PAH and VN-PAH patients. This figure focuses on the Wnt signaling pathway. Note the differing frequency of gene mutations as well as the differences in novel gene variants between VR-PAH and NR-PAH. Reproduced with permission of the American Thoracic Society, from Hemnes et al.
10
Copyright © The American Thoracic Society. The American Journal of Respiratory and Critical Care Medicine is an official journal of the American Thoracic Society.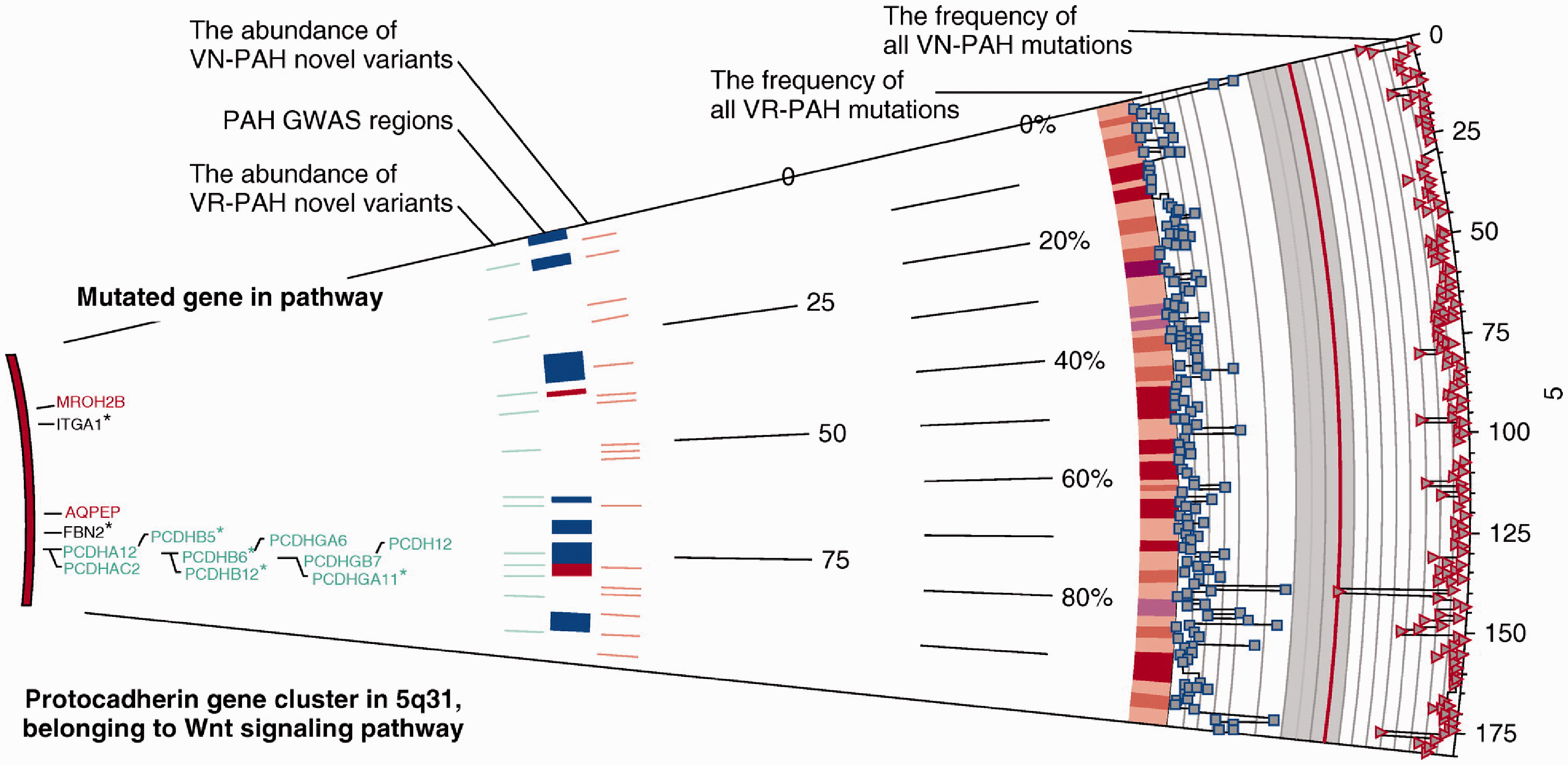
There have been no recent descriptions of pulmonary vascular histology in responders to an acute vasodilator challenge or in long-term calcium blocker responders. These patients have a lengthy survival and have not presented to autopsy. Given the rapid dramatic reduction in vascular resistance during acute vasodilator challenge in responders, one hypothesis is that the problem resides in the precapillary arteriolar smooth muscle cell layer and results nearly exclusively from vasoconstriction, and that the intimal cellular proliferation seen in many patients with IPAH must not be predominant. In support of this, the lung tissue from a ten-year-old boy with IPAH and significant acute vasodilator responsiveness showed only medial hypertrophy and minimal intimal thickening in the pulmonary arteries. 67 Moreover, in the only morphometric study of lung biopsy tissue from VR-PAH, defined as a >30% decrease in PVR and a >10% reduction in mPAP, the presence of intimal cellular thickening with an intimal area > 18% of vascular cross-sectional area, had an 85% predictive value for VN-PAH. 68
The distinct physiology of VR-PAH
A recent study has demonstrated the distinct physiology of VR-PAH versus VN-PAH, identifies the difference between “resistance” and “pressure” responders during an acute vasodilator challenge, and confirms that an isolated decrease in PVR in the absence of decreased PAP is not beneficial.8,69 It also provides evidence that the VR-PAH lung can “normalize” perfusion with therapy, possibly explaining the excellent long-term prognosis for treated VR-PAH. Using a technique that measures first-pass transpulmonary metabolism (%M) and hydrolysis (v) of 3H-Benzoyl-Phe-Ala-Pro (BPAP) by the pulmonary capillary endothelium-bound angiotensin-converting ectoenzyme (ACE), the perfused functional capillary surface area (FCSA) can be determined and normalized to body surface area (FCSA/BSA).70–74 The technique enables differentiating increased precapillary blood flow leading to microvascular recruitment, from increases in flow that simply distend already perfused vessels.75,76 Twelve NR-PAH and two VR-PAH patients were studied, pre and at peak tolerated dose of intravenous epoprostenol (Fig. 7). The VR-PAH patients met the current criteria for vasoresponsiveness and were true “pressure” responders.
50
In the VN-PAH group, cardiac output increased by an average of 36%, with no change in mPAP, and an average decrease in PVR of 31%, typical of a “resistance” response. Determination of FCSA/BSA revealed reduced baseline levels in NR-PAH, indicative of an underperfused capillary bed with flow restricted by a remodeled upstream arteriolar bed. Moreover, despite increased cardiac output with the epoprostenol infusion, there was no recruitment of FCSA in the VN-PAH group, indicating persistent upstream pulmonary arteriolar luminal obstruction. Instead, the increased cardiac output was accommodated simply by distention of already perfused capillaries, with a shorter transit time, as evidenced by the decrease in %M and unchanged FCSA in many patients.
76
These findings provide a physiological basis for the “resistance” response and may explain the clinical failure of vasodilators that only provided a reduction in PVR without decreasing PAP.
Changes in mPAP, cardiac output, PVR, percent metabolism of 3H-BPAP, hydrolysis of BPAP (v), and determination of functional capillary surface area (FCSA) normalized to body surface area (BSA), before (Pre) and at peak (Peak) dose of an acute vasodilator challenge in patients with IPAH. The unfilled circles represent VN-PAH, the filled circles represent two patients with VR-PAH, and the filled gray square and triangle represent two VN-PAH patients with hemodynamic abnormalities of a similar severity to the patients with VR-PAH. Only the VR-PAH patients successfully recruit FCSA during the vasodilator challenge. Data are from Langleben et al.
8
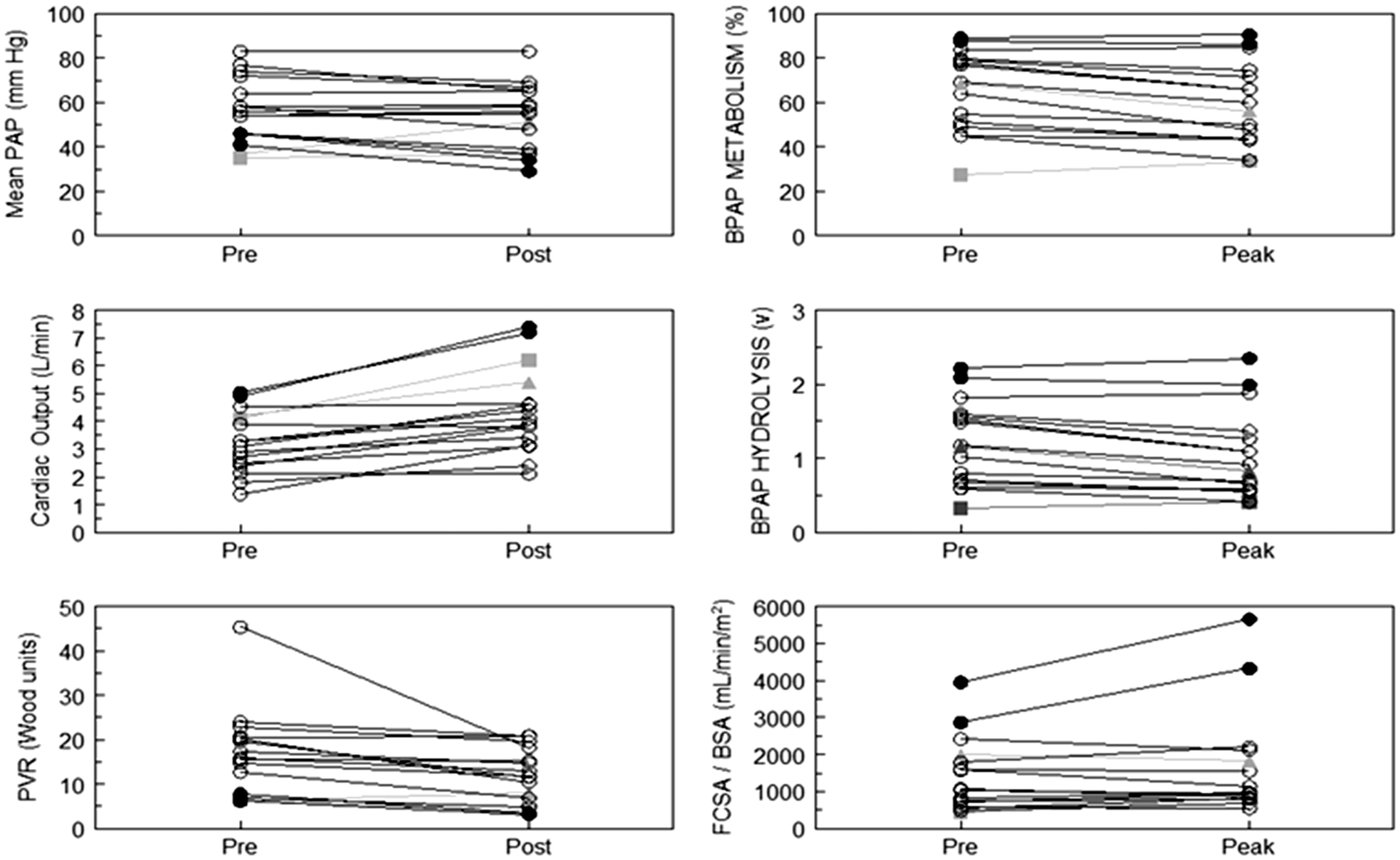
By contrast, the VR-PAH participants had a true “pressure” response, with decreased mPAP and increased cardiac output, resulting in decreased PVR. In those patents, FCSA/BSA was higher at baseline, but increased dramatically with epoprostenol. The rise in FCSA/BSA with little or no change in %M indicates that the increased cardiac output from rapid vasodilation of precapillary arterioles was accommodated via capillary recruitment, rather than distention. 76 The recruitment response seen in the VR-PAH patients is identical to that seen in normal humans performing exercise. It was noted previously that VR-PAH patients may have milder pulmonary hypertension at the time of evaluation. 7 However, comparing the two VR-PAH with two VN-PAH that also had mild hemodynamic abnormalities, it is clear that the VN-PAH do not recruit microvasculature even when baseline hemodynamic abnormalities are mild (Fig. 7).
Given the rapid vasodilator response during an acute vasodilator challenge, these findings strongly suggest different microvascular abnormalities between VR-PAH and VN-PAH. Patients with VR-PAH must have principally and nearly exclusively a vasoconstrictive abnormality (Fig. 8). The acute vasoreactivity does not imply complete resolution of the vascular abnormality, only a reduction of the resistance to hemodynamically insignificant levels. By contrast, most VN-PAH patients must have severe cellular narrowing of arterioles, representing a physical obstruction to flow into underperfused capillary areas. Given that the distribution of histologic abnormalities is not uniform in IPAH, there may be some regions of the lung that can never see increased perfusion despite a higher driving cardiac output and the already, albeit abnormally, perfused regions must carry the flow via distention.
Schematic (not anatomic, nor positional) illustrations of pulmonary capillary perfusion in VR-PAH and VN-PAH. The ovals represent only a potential amount of capillary perfusion and do not represent acini. The blue vessels represent pulmonary arteries feeding into the capillaries. Pulmonary venous drainage is not shown. Left: VR-PAH, prior to vasodilation. Precapillary constriction increases PVR and reduces capillary perfusion diffusely; center: VR-PAH, during successful vasodilation. A marked increase in capillary perfusion occurs; right: VN-PAH. The precapillary arterioles have intraluminal cellular obstruction that greatly reduces or eliminates capillary perfusion. Any increased pulmonary blood flow during the acute vasodilator challenge is accommodated via distention of perfused capillaries, and not via recruitment.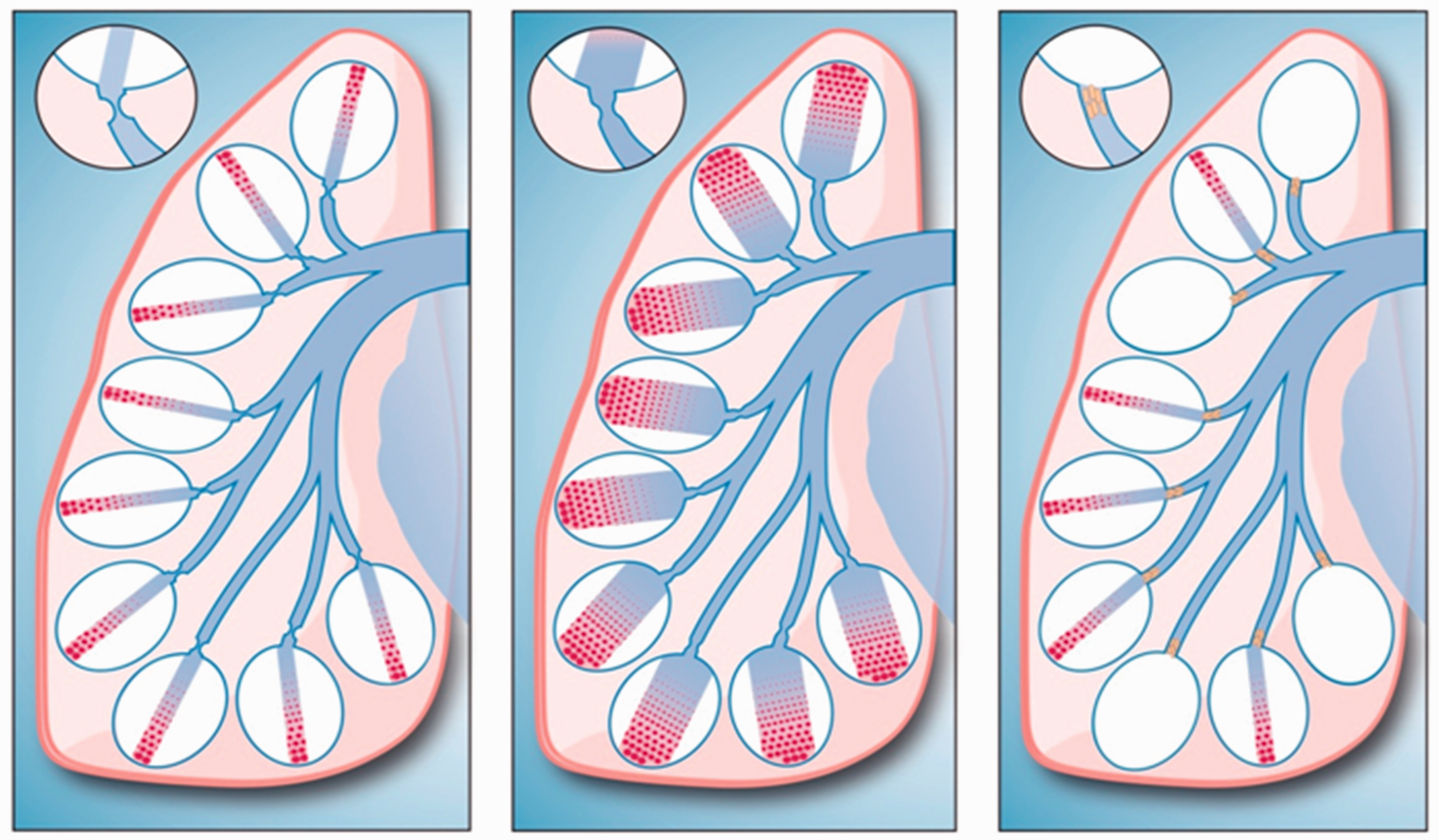
The most encouraging finding in the study was the normalization of FCSA in the VR-PAH patients. This supports the long-term improvement in hemodynamics seen in those patients and the excellent prognosis. In support of this, a recent nuclear imaging study, using a radiolabeled adrenomedullin derivative, assessed pulmonary blood flow patterns in PAH patients.
77
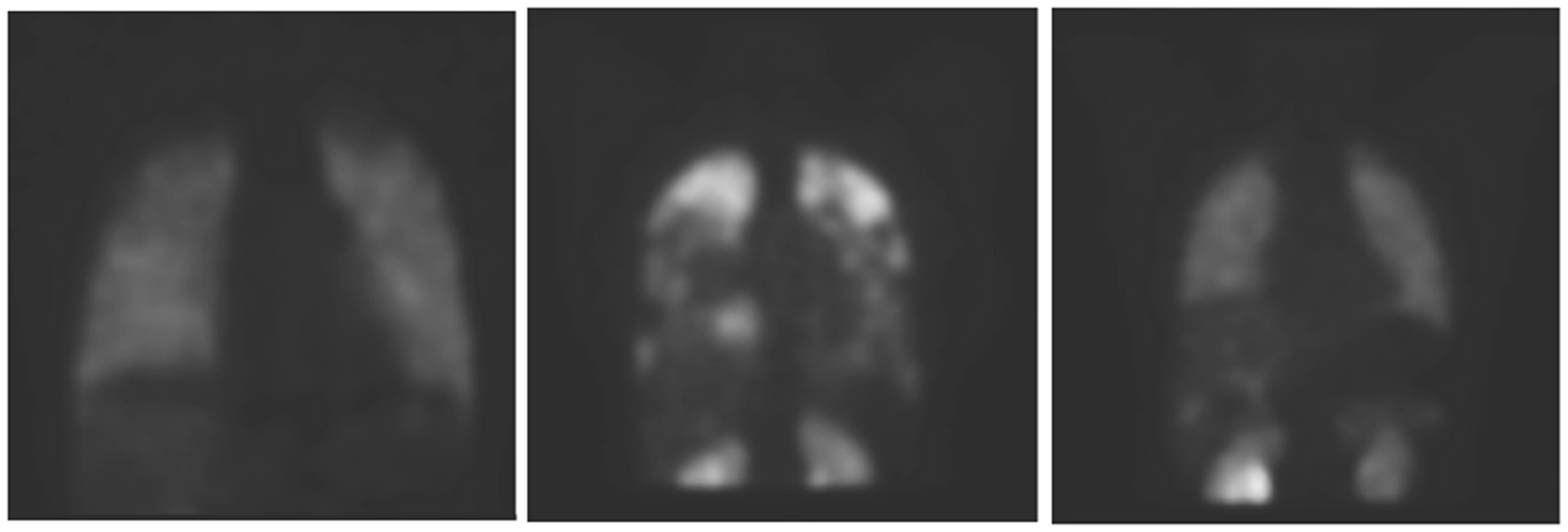
It showed a completely normal perfusion pattern in one VR-PAH patient on calcium blockers, compared with persistently abnormal perfusion patterns in VN-PAH patients treated with approved PAH therapies (Fig. 9).
Radionuclide lung scans, using a molecule that binds to the endothelial adrenomedullin receptor, 99mTc-PulmoBind. Left: normal human; center: NR-PAH; right: VR-PAH on high dose calcium blocker therapy. Note the normal pattern of perfusion in VR-PAH as opposed to NR-PAH. Reproduced with permission from Harel et al.
77
Conclusion
There is now strong molecular, physiologic, and clinical evidence that true acute vasodilator responsive IPAH is a distinct clinical entity, with a specific therapy that provides long-term remissions. Efforts should be ongoing to provide a non-invasive method of diagnosing it and to provide a complete understanding of its pathogenesis. The fundamental problem may lie in abnormalities of cellular or mitochondrial calcium handling, in energetics of ion transport, or in other as yet unforeseen mechanisms. Further genome-wide studies should aid greatly and their chance of success will increase through multicenter collaborations that provide a larger cohort of carefully characterized VR-PAH patients. The progress in molecular characterization of VR-PAH, although incomplete, suggests that precision medicine is possible in PAH, offering the hope that it will lead to more effective therapy for VN-PAH. The fact that normal or near-normal microvascular perfusion can be attained with appropriate therapy in VR-PAH offers the hope that a similar objective can be reached in VN-PAH, when the precise therapies for that condition are found. Consideration should be given to assigning VR-PAH a distinct category within future classifications of group 1 pulmonary hypertension.
Footnotes
Conflict of interest
The author(s) declare that there is no conflict of interest.
Funding
This study was supported in part by the William and Ida Pencer Family Foundation, the Dimitrios Banousis Foundation, the Bank of Montreal Center for the Study of Heart Disease in Women, and the JGH Annual Walk for Pulmonary Hypertension, all at the Jewish General Hospital.