
Abstract
Pulmonary arterial hypertension (PAH) is a complex disease with a poor prognosis. Selexipag is a selective prostacyclin receptor agonist with vasodilatory, anti-proliferative, anti-inflammatory, and pro-angiogenic properties. However, no clinical data on its therapeutic use in children with PAH are currently available. Here, we report the case of a 12-year-old girl who presented in World Health Organization (WHO) functional class III and right ventricular (RV) failure with recurrent syncope, dizziness, and progressive fatigue for two years. Cardiac catheterization revealed severe precapillary PAH: mean right atrial pressure (RAP) = 10–13 mmHg, right ventricular end-diastolic pressure (RVEDP) = 13 mmHg, left ventricular end-diastolic pressure (LVEDP) = 7 mmHg, mean pulmonary arterial pressure (PAP) = 81 mmHg, and mean aorta ascendens pressure = 89 mmHg. The pulmonary vascular resistance index (PVRi) was 25.2 WU × m2. An oral combination therapy was started with a phosphodiesterase type 5 inhibitor (sildenafil 3 × 20 mg) and an endothelin-1 receptor antagonist (bosentan 2 × 62.5 mg). No significant clinical/hemodynamic improvement was seen after nine months of dual therapy, so that the patient was transferred to our institution. We agreed upon the off-label add-on use of oral selexipag. Within ten days, we up-titrated selexipag to a final (max. adult) dose of 1600 mcg twice daily. After six months, the patient had: (1) decrease in PVR index, pulmonary artery acceleration time, RAP, RVEDP, right atrial/RV size; (2) re-gain of vasoreactivity; and (3) improvement of cardiac index, 6-minute walking distance, functional class, body weight, and CAMPHOR score. Our encouraging results suggest the consideration of off-label use of oral selexipag in children with severe PAH, preferably in a protocol-driven prospective study.
Pulmonary arterial hypertension (PAH) is a complex disease with a poor prognosis despite major improvements in pharmacotherapy over the last two decades. PAH is thought to be initiated by endothelial damage and is associated with an excessive production of growth factors, pro-inflammatory mediators, and vasoconstrictors (endothelin-1, thromboxane), a fade of vasodilators such as prostacyclin and nitric oxide, and ultimately obliterative pulmonary vascular remodeling and right ventricular (RV) failure. 1 Today, PAH-targeted therapy addresses three major pathways: endothelin-1, nitric oxide, and prostacyclin. 2 Moreover, many PAH centers use off-label drugs for compassionate use in adults and children. Selexipag (syn. NS-304, ACT-293987) is a selective prostacyclin (IP) receptor agonist 3 that has not only vasodilatory but also anti-proliferative properties, anti-inflammatory, and pro-angiogenic properties. 1 A placebo-controlled phase 3 trial demonstrated that selexipag, at a maximum dose of 1600 mcg twice daily, significantly reduced the risk of the composite primary end point of death or PAH-related complication versus placebo in adults (GRIPHON, n = 1156; heart rate [HR] = 0.60; 99% confidence interval [CI] = 0.46–0.78; P < 0.001). 4 Thus, selexipag appears to offer a promising oral agent targeting the prostacyclin pathway. However, no clinical data on its use, titration, adverse effects, and efficiency in children with PAH are currently available.
A 12-year-old girl (weight = 27.7 kg, <3rd percentile (P3), height = 136 cm, <P3; BMI = 14.7 kg/m2) presented in September 2015 with recurrent syncope, dizziness, and progressive fatigue which had started two years ago (World Health Organization [WHO] functional class III). On echocardiography, she had a dilated and hypertrophied RV. The estimated RV systolic pressure was 100 mmHg. A chest computed tomography (CT) with pulmonary artery angiography excluded an underlying parenchymal lung disease, pulmonary veno-occlusive disease, chronic thromboembolic pulmonary hypertension, and anatomical obstructions. Cardiac catheterization revealed an increased mean right atrial pressure (mRAP) of 10–13 mmHg, a RV end-diastolic pressure (RVEDP) of 13 mmHg, a left ventricular end-diastolic pressure (LVEDP) of 7 mmHg, a mean pulmonary artery pressure (PAP) of 81 mmHg (systolic PAP = 103 mmHg, diastolic PAP = 64 mmHg), and a mean aorta ascendens (AAO) pressure of 89 mmHg (systolic = 105 mmHg, diastolic = 74 mmHg). The pulmonary vascular resistance (PVR) index was calculated to be 25.2 Wood units (WU) × m2 resulting in a PVR/systemic vascular resistance (SVR) ratio of 1.2. At this first catheterization, the patient was responsive to vasodilators (oxygen, nitric oxide, and Iloprost) with a 50% reduction in PVR index. Laboratory tests showed no abnormalities except for low serum ferritin. Serum B-type natriuretic peptide concentration was 76.2 pg/mL (reference range = 0–89) at presentation. Based on the above findings (WHO functional class III, clinical evidence of RV failure with progression of symptoms including syncope, severe RV enlargement, PVR index > 15 WU × m2), the girl was stratified as a high-risk PAH patient. 5 An oral upfront PAH combination therapy was started with a phosphodiesterase type 5 inhibitor (sildenafil 3 × 20 mg), endothelin-1 receptor antagonist (bosentan 2 × 32 mg), and anticoagulant (warfarin, INR 2–2.5). Six weeks after first diagnosis, bosentan was increased to 2 × 62.5 mg/d because of progressive complaints of fatigue. Six months later the girl was in a so-called “stable” condition (no recurrent syncope) but still in WHO functional class III. She underwent a second cardiac catheterization to re-evaluate the effect of pharmacotherapy. There was no improvement of hemodynamics and loss of responsiveness to acute vasodilator testing. The patient was then referred to our PAH center for further diagnostics and advanced therapies. We found a missense mutation in the activin receptor-like kinase 1 gene (ACVRL1; syn. ALK-1), i.e. a TGFβ superfamily, classifying her to have hereditary rather than idiopathic PAH. She was lacking any oro-mucal and intrapulmonary features of hemorrhagic telangiectasia type 2 that is known to be associated with ACVRL1 mutations.
We discussed escalating PAH-targeted drug therapy since this patient was in WHO functional class III, and strongly considered parenteral prostacyclin analogues (PCA) for combined triple therapy. However, due to the patient’s low body weight (30 kg, +2.3 kg in six months but still <P3), the implantation of a subcutaneous pump for intravenous administration of the PCA trepostinil was not found to be a suitable option at this stage. We therefore agreed upon the off-label add-on therapy of oral selexipag with the young teenager and her caregiver, and discontinued warfarin, which we do not use routinely in children with PAH. Within ten days, we up-titrated oral selexipag to a final (max. adult) dose of 1600 mcg twice daily, starting with 200 mcg in the evening and increasing the single dose by 200 mcg every 24 h. Except for mild to moderate nausea that improved with food intake, no severe adverse effects were observed, e.g. jaw pain. Two weeks after starting selexipag add-on drug therapy, she was discharged home with stable vital signs (HR = 71 bpm, blood pressure = 100–107/64–68 mmHg), on sildenafil, bosentan, selexipag, spironolactone, and the antiemetic ondansetron. Clinically, and by echocardiography, the patient started to improve remarkably within the first two to three months after initiation of selexipag. After six months, we conducted a full clinical and hemodynamic re-evaluation, including right and left heart catheterization with encouraging results: (1) decrease in all, PVR index, pulmonary artery acceleration time, mean right atrial (RA) pressure, RVEDP, RA and RV dilation; (2) re-gain of vasoreactivity; and (3) improvement of cardiac index, 6-minute walking distance, WHO functional class, body weight, and CAMPHOR clinical self-assessment score (Fig. 1).
Hemodynamic and clinical improvement in a 12-year-old girl with severe PAH during the first six months after reaching the target dose of oral selexipag, as add-on to background therapy (sildenafil, bosentan). Decrease in PAP and transpulmonary pressure gradient (TPG), measured by cardiac catheterization six months after start of add-on oral selexipag treatment (a). Decrease in RVEDP and mean RAP and slight increase of CI, measured by cardiac catheterization six months after start of selexipag treatment (b). Decrease of PVR index as measured by cardiac catheterization six months after start of selexipag treatment (c). Improvement of right ventricular dilation (RV/LV end-systolic ratio) and left ventricular compression (LV end-systolic eccentricity index) as demonstrated on transthoracic echocardiography three and six months after start of selexipag treatment (d). Increase in PAAT measured by PW-Doppler in the parasternal short axis view, three and six months after start of selexipag treatment (e). Reduction in RA size measured by echocardiography from the four-chamber view, three and six months after start of selexipag treatment (f). Improvement of the 6-minute walking distance three and six months after start of selexipag treatment (g). Reduction of symptoms, physical impairment, and quality of life impairment, as reported by self-assessment using the Cambridge Pulmonary Hypertension Outcome Review (CAMPHOR) questionnaire before and six months after start of oral selexipag therapy. (h) CAMPHOR, Cambridge Pulmonary Hypertension Outcome Review Questionnaire; CATH, cardiac catheterization; CI, cardiac index; dPAP, diastolic pulmonary arterial pressure, dTPG, diastolic transpulmonary pressure gradient; Ecc. Index, eccentricity index; ECHO, echocardiography; LV, left ventricle; mPAP, mean pulmonary arterial pressure; mRAP, mean right atrial pressure; PA, pulmonary artery; PAAT, pulmonary artery acceleration time; PVR, pulmonary vascular resistance; QoL, quality of life; RA, right atrium; RV, right ventricle; RVEDP, right ventricular end-diastolic pressure; sPAP, systolic pulmonary arterial pressure; WU, Wood units.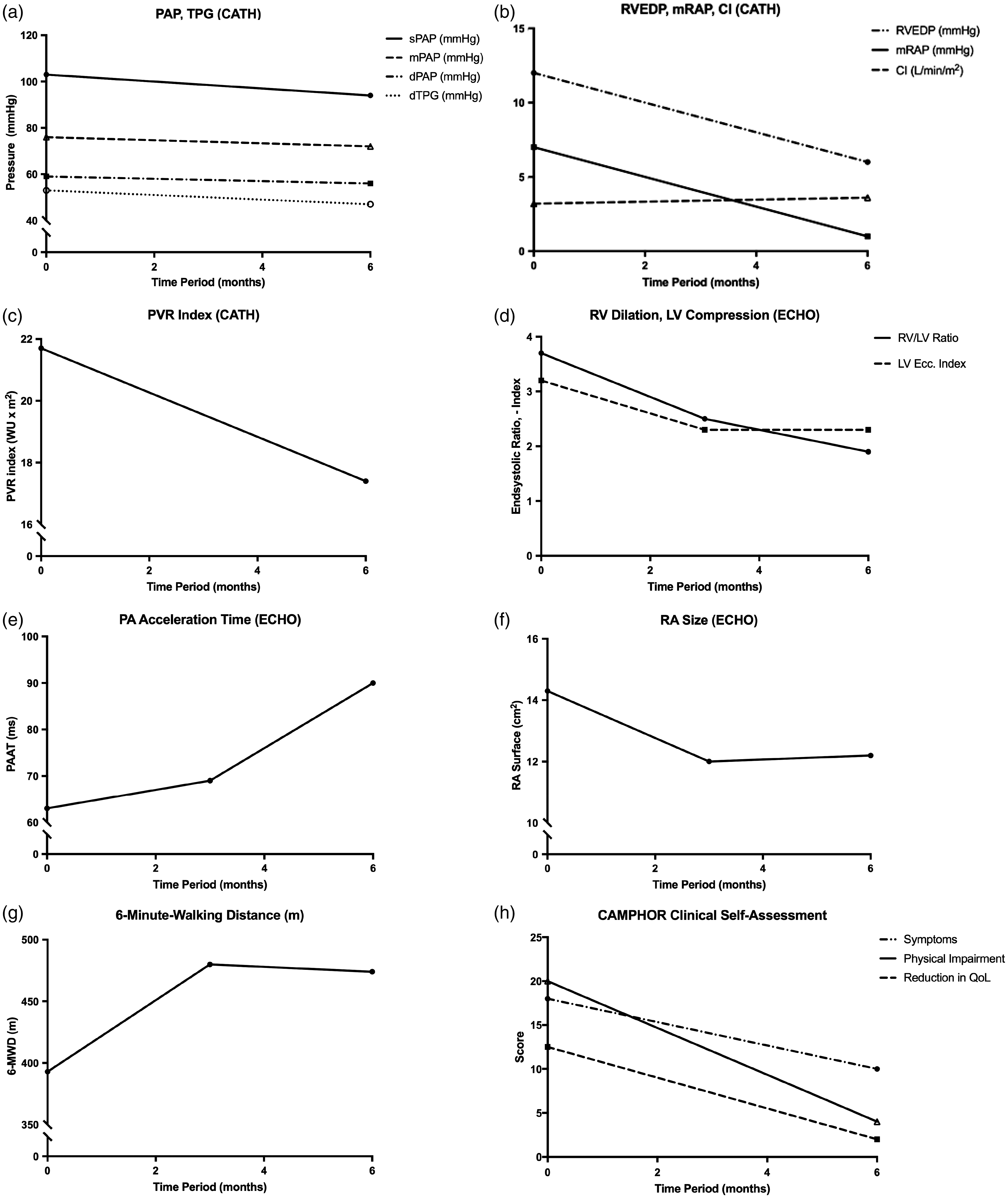
To the best of our knowledge, this is the first report on the use of oral selexipag for severe, systemic PAH and associated RV failure in childhood. Except for mild to moderate nausea, selexipag was well tolerated. Clinically, the teenager improved remarkably on selexipag at short-term follow-up, along with improvement of several prognostic hemodynamic and anatomic variables. Meanwhile, a few PH centers have started add-on selexipag in a limited number of children with severe PAH (U. Kanaan, Atlanta; M. Koestenberger, Graz; personal communication), while others are awaiting more preliminary clinical data or the opportunity to enroll their patients into a clinical trial. Our first report provides important information on drug dosing, management of adverse effects, and the hemodynamic and clinical effects of selexipag that may be achieved within six months of treatment.
Footnotes
Conflict of interest
Funding
GH currently receives grant support from the German Research Foundation (DFG; HA 4348/2-1, 4348/6-1), Kinderherzen e.V. (W-H-001-2014), and Stiftung Kinderherz (2511-6-13-011).