
Abstract
Postpartum depression is considered to be a subtype of major depressive disorder that occurs in approximately 10–20% of mothers worldwide. However, in actuality, these numbers are likely underreported due to minimization and the stigma of mental illness. Until recently, there were no approved medications for the treatment of postpartum depression. Allopregnanolone is a naturally occurring neuroactive steroid whose serum levels decline precipitously following childbirth. This hormonal fluctuation has been postulated as playing a role in the pathophysiology of postpartum depression. Brexanolone is the first medication approved by the US Food and Drug Administration for the treatment of postpartum depression. Brexanolone is an intravenous proprietary formulation of allopregnanolone that can be administered to produce stable serum levels comparable with third-trimester concentrations in postpartum mothers. It is hypothesized to modulate neuronal excitability by functioning as an allosteric modulator of γ-aminobutyric acid-A receptors and is administered under monitoring as a 60 h continuous infusion. In this review, we will highlight the results of the clinical trial program, including efficacy and tolerability data. Practical and logistical considerations of brexanolone will be reviewed, as will its potential place in therapy for the treatment of postpartum depression.
Introduction
Diagnosis
Depression with peripartum onset, otherwise known as postpartum depression (PPD), is defined by the American Psychiatric Association (APA) as a depressive episode occurring during the third trimester of pregnancy or in the 4 weeks following delivery. 1 The American College of Obstetricians and Gynecologists (ACOG) provides a more expansive definition, considering PPD to be a major or minor depressive episode that occurs during pregnancy or in the first 12 months after delivery. 2 The depressive episode must cause significant distress or impairment in function. Upwards of 50% of postpartum depressive episodes are felt to begin prior to delivery, giving credence to the term peripartum depression. 3 Phenomenologically, PPD is considered a subtype of major depressive disorder (MDD). Symptoms that typically distinguish PPD from MDD include severe anxiety, panic attacks, and ruminations, which can manifest as the primary complaint.4,5
Prevalence and consequences
PPD occurs in approximately 10–20% of mothers worldwide, with a comparable prevalence of 8–20% in the USA.6 –9 However, these numbers are likely underreported due to the stigma associated with mental illness, and the minimization that occurs from both mothers and healthcare providers.10,11 Evidence suggests that education, race, and ethnicity can further impact the self-under-reporting of PPD. 12 A low mood (often called ‘baby blues’) is common in the postpartum period and can affect approximately 50% of mothers, but this is usually mild and transient, does not meet criteria for a major depressive episode and is not a formal psychiatric diagnosis; postpartum depression extends in symptom severity beyond this. 13 It is possible that the stigma of diagnosing PPD, even when justified, can lead to a further under-reporting of the condition, and normalizing it as ‘baby blues’.
Mothers with PPD have an increased risk of morbidity and mortality, with one in seven deaths in the first postpartum year being a result of suicide.14 –16 However, the consequences of PPD are far-reaching and extend beyond the impacted mother. Afflicted mothers show an overall decrease in attention to infant needs, causing a delay in the child’s height, weight, and emotional and behavioral development.17,18 Disrupted and insufficient mother–child attunement can also result in impaired child social and interpersonal functioning. These effects can last throughout childhood and adolescence. In a study of 9848 mothers assessed for PPD, PPD was found to double the risk of child behavioral disturbances [moderate PPD odds ratio (OR) was 2.22 (95% confidence interval (CI) 1.74–2.83), severe PPD was 1.91 (95% CI 1.36–2.68)] manifesting at 3.5 years of age (OR 4.84; 95% CI 2.94–7.98). Maladaptive outcomes persisted, and children had lower mathematics grades at age 16 (OR 2.65; 95% CI 1.26–5.57), and higher rates of depression at age 18 (OR 7.44; 95% CI 2.89–19.11). 19 In the most severe of cases, PPD has also been linked to infanticide, underscoring the critical importance of prompt recognition and treatment. 20
Consensus guidelines recommend screening for depression during pregnancy and the postpartum period.2,21,22 In the USA, some states mandate the screening of PPD prior to discharge from the birthing hospital and at the first few postpartum checkups. 23 The most commonly used screening test among obstetricians is the 10-item Edinburgh Postnatal Depression Scale (EPDS). 24 Among many primary care providers, the Patient Health Questionnaire (PHQ-9) is generally used for all patients to screen for all types of depression. 25
In those with a history of PPD, the risk of a PPD in future pregnancies is estimated to be 27–46 times higher than in those without prior PPD. 26 For some patients, the relationship between PPD and MDD may also be analogous to the relationship between gestational diabetes and type 2 diabetes. Gestational diabetes typically goes away after childbirth; however, the risk of subsequently developing type 2 diabetes is up to seven times higher than in women who had a normoglycemic pregnancy. 27 Similarly, PPD increases the risk of future non-perinatal depressive episodes; however, a caveat is that many will not have a recurrence outside of a pregnancy or peripartum period. 26
Etiology
The cause of PPD is complex and likely multifactorial, with several possible etiologies ranging from environmental to hormonal to neurotransmitter dysfunction. A genetic predisposition with genetic polymorphisms is another possibility. 28 A focus of current interest is on the interplay between the hypothalamic–pituitary–adrenal axis (HPAA), hormonal fluctuations, and γ-aminobutyric acid (GABA) signaling during the peripartum and postpartum period. 29 Allopregnanolone, a neuroactive steroid and endogenous progesterone metabolite synthesized in the central nervous system, is felt to modulate neuronal excitability by acting on the GABA-A receptor as a positive allosteric modulator. 30 In rodent and human models, neuroactive steroids have been established as potential therapeutics for PPD related to their ability to modulate the GABA-A receptor. Deficiencies in functional GABA signaling results in an inability to suppress stress-induced activation of the HPAA, a potential causative factor for PPD.31,32 Allopregnanolone concentrations rise throughout pregnancy and reach peak concentrations during the third trimester. 33 Following childbirth, allopregnanolone levels decrease precipitously. 34 Failure of GABA-A receptors to adapt to changing hormonal levels has been hypothesized as a factor for precipitating PPD.35 –37 In a double-blind randomized control trial (RCT), simulating the effects of hormonal fluctuation that occurs during parturition was shown to precipitate depression in women with a history of PPD but not in those without. 38
Existing treatments
Systematic reviews have been conducted to investigate the efficacy of antidepressants and psychotherapies for the treatment and prevention of PPD.39 –43 These studies have yielded mixed results, highlighting limited data and the small number of available studies. None of the available antidepressants indicated for the treatment of MDD have received regulatory approval specifically for PPD. Moreover, some data suggest that there is a greater time to treatment response, with lower rates of remission, with these treatments in perinatal women than in non-perinatal women with MDD.44,45 A possible explanation for these findings is that the commonly used antidepressants targeting monoamines are not directly linked with the hormonal hypothesis behind the etiology of PPD. Identifying treatments with a rapid onset of action is needed to mitigate the negative sequalae of PPD on both the mother and family.
The focus of this review will be on brexanolone intravenous infusion, a ‘first in class’ novel medication for the treatment of PPD. Brexanolone was approved for the treatment of PPD in adults by the US Food and Drug Administration (FDA) in 2019. 46 A literature search was conducted on 11 April 2020 using the US National Library of Medicine’s PubMed.gov resource (https://www.ncbi.nlm.nih.gov/pubmed/) using the words ‘sage-547’ OR ‘brexanolone’. Only full-text articles were considered and there were no other filters or constraints. The search yielded a total of 51 results, and each publication was reviewed by the authors to determine if data pertaining to the efficacy and/or tolerability of brexanolone was provided.
Brexanolone pharmacology
During pregnancy, there is a rise in allopregnanolone levels before an abrupt decrease during childbirth. Fluctuations in allopregnanolone as well as GABAergic signaling in the peripartum period may play a role in the development of PPD. PPD occurs at different times in different women; some during pregnancy when allopregnanolone levels are high, and others during the post-partum period following the drop in allopregnanolone. It is not known whether these two groups of women are similar or different; however, both groups coincide with changes in the duration and amplitude of neuroactive steroid exposure, giving credence to exploring allopregnanolone for the treatment of PPD. 47 Allopregnanolone has poor oral bioavailability and is rapidly metabolized. However, brexanolone (formerly sage-547), a proprietary formulation, is an aqueous β-cyclodextrin-based formulation of allopregnanolone that can be administered intravenously to produce stable serum levels and is chemically identical to naturally made allopregnanolone. 47 How brexanolone functions in the treatment of PPD is not entirely understood, but is thought to be related to GABA-A receptor activity. To compensate for the precipitous decline in allopregnanolone that naturally occurs immediately following childbirth, brexanolone provides postpartum mothers with doses of allopregnanolone comparable with endogenous third-trimester serum concentrations. 48 Tapering allopregnanolone (by utilizing brexanolone) may give postpartum mothers additional time for physiologic adaptation to the decreasing allopregnanolone levels, and reduce the possibility of adverse events such as worsening anxiety or panic attacks because of brexanolone’s rapid modulation of the GABA-A receptor. 48 GABA-A receptors may adapt better to gradual tapering rather than an abrupt decrease in allopregnanolone levels that occurs at parturition, preventing a shift in the excitatory–inhibitory balance in the brain.35,49 Treatment with brexanolone involves a regimented and continuous intravenous infusion over the course of 60 h: 30 µg/kg per hour (0–4 h), 60 µg/kg per hour (4–24 h), 90 µg/kg per hour (24–52 h), 60 µg/kg per hour (52–56 h), 30 µg/kg per hour (56–60 h). 50
Brexanolone is greater than 99% protein bound and has a terminal half-life of 9 h. It is not metabolized through the cytochrome P450 system but rather through ketoreduction, glucuronidation, and sulfation. Brexanolone is excreted through feces (47%), primarily as metabolites, and urine (42%), with less than 1% unchanged. There are no known drug–drug interactions, and no hepatic contraindications. There are no renal contraindications for mild, moderate, or severe renal impairment, but brexanolone should be avoided in those with end-stage renal disease. 50
Efficacy
The efficacy and tolerability of brexanolone was studied in women suffering from a unipolar major depressive episode with peripartum onset and was not tested in women with bipolar disorder who had a peripartum onset major depressive episode. Drug infusions all took place at inpatient hospitals or at research centers where the patients stayed for the duration of 72 h. No outpatient infusion centers were utilized. The efficacy of brexanolone has been established in one proof-of-concept trial, one phase II trial, and two phase III trials. The primary efficacy outcome measure for each RCT was the change from baseline on the 17-item Hamilton Depression Rating Scale (HAM-D) at 60 h in patients who received the study drug or placebo.29,47,51 In an open-label proof-of-concept trial, four patients admitted to an inpatient psychiatric unit for a major depressive episode that were no earlier than the third trimester of pregnancy, and no later than 12 weeks postpartum, received a continuous intravenous (IV) infusion of brexanolone for 60 h. 48 Mean HAM-D total score at baseline was 26.5 ± 4.1, and the mean HAM-D total score was 1.8 ± 1.5 (p = 0.001 versus baseline) at hour 60 and 5.3 ± 2.9 at hour 84 (81% decrease from baseline at hour 84). Mean Clinical Global Impression-Improvement (CGI-I) score was 1.8 ± 0.5 at hour 12 and remained <2 at each subsequent assessment. 52
In the phase II study, 21 ambulatory patients with a major depressive episode that began no earlier than the third trimester, and no later than the first 4 weeks following delivery, were enrolled and randomly assigned to receive treatment with brexanolone 90 µg/kg per hour (BRX90; n = 10) or placebo (n = 11). 47 Patients were ⩽6 months postpartum with severe PPD (HAM-D score ⩾26). Patients in the brexanolone group received 30 µg/kg per hour (0–4 h); 60 µg/kg per hour (4–24 h); 90 µg/kg per hour (24–52 h); 60 µg/kg per hour (52–56 h); 30 µg/kg per hour (56–60 h). On the primary endpoint, the mean change from baseline at 60 h was −20.97 in the brexanolone group and −8.75 in the placebo group with a mean difference between groups of −12.2 points (95% CI −20.77 to −3.67; p = 0.0075). There was an effect size for clinical efficacy of 1.2. Secondary endpoints were not adjusted for multiplicity. On secondary endpoints, a statistically significant difference between the brexanolone group and placebo group was met at 24 h [−19.37 and −8.12, respectively (p = 0.0059)], and this statistical significance between brexanolone and placebo persisted at all subsequent measurements (36 h, 48 h, 60 h, 72 h, 7 days, 30 days). HAM-D results were consistent with change from baseline on the Montgomery–Åsberg Depression Rating Scale (MADRS), with a mean difference between groups at 24 h of −17.5 (p = 0.0042), −15.9 (p = 0.01) at 60 h, and −15.1 (p = 0.01) at 30 days. 53
On other secondary endpoints, remission from depression (HAM-D total score ⩽7) was seen in: (a) 6 of 10 patients in the brexanolone group, and 1 of 11 patients in the placebo group at 24 h (OR 15.00; 95% CI 1.07–756.72; p = 0.0561); (b) 7 versus 1 patient at 60 h (OR −23.33; 95% CI −1.56 to 1152.71; p = 0.0364); and (c) 7 versus 2 patients at 30 days (OR 10.50; 95% CI 1.01–140.57; p = 0.0499). 48 More patients in the brexanolone group achieved treatment response, defined as ⩾50% reduction in HAM-D total score, at all timepoints, though it was not significant at 60 h (seven patients in the brexanolone group versus four patients in the placebo group). Improvements in Columbia-Suicide Severity Rating Scale suicidal ideation items were noted in both the brexanolone and placebo groups. 54
Two phase III, double-blind, randomized, placebo-controlled studies were conducted by Meltzer-Brody and colleagues. 29 Study participants consisted of ambulatory females aged 18–45 in good physical health, having a major depressive episode, with onset no earlier than the third trimester and no later than 4 weeks after delivery, with a qualifying HAM-D total score (⩾26 for study 1; 20–25 for study 2) before infusion, and were ⩽6 months postpartum at screening. Patients were randomly assigned (1:1:1) to receive brexanolone 90 µg/kg per hour (BRX90), brexanolone 60 µg/kg per hour (BRX60), or matching placebo for 60 h in study one, or (1:1) BRX90 or matching placebo for 60 h in study two. Across both studies, 22% of patients were taking antidepressants at baseline. Primary and secondary endpoints were consistent between studies and with the phase II study.
In study one, 138 patients were randomized to receive BRX90 (n = 45), BRX60 (n = 47), or placebo (n = 46). On the primary endpoint, the change from baseline in HAM-D total score at the end of the 60 h infusion was −17.7 [standard error of the mean (SE) 1.2], −19.5 (SE 1.2), and −14.0 (SE 1.1) in the BRX90, BRX60 and placebo groups, respectively. The mean change in both brexanolone groups was statistically significantly different from the placebo group [−3.7 (95% CI −6.9 to −0.5), p = 0.0252 for BRX90; −5.5 (−8.8 to −2.2), p = 0.0013 for BRX60]. The improvement in HAM-D total score from baseline observed at 60 h was sustained at day 30 [BRX90 −17.7 versus −17.6 (SE 1.4); BRX60 −19.5 versus −19.5 (SE 1.4); placebo −14.0 versus −13.8 (SE 1.3)]. There was a statistically significant (p = ⩽0.05) difference in the change from baseline HAM-D scores between BRX60 and placebo at 24 h, and all subsequent measurements (36 h, 48 h, 60 h, 72 h, 7 days, 30 days); however, BRX90 was only significantly different from placebo at 60 h and 30 days. The proportion of patients who achieved HAM-D response was higher for both brexanolone groups when compared with placebo, with statistical significance achieved across multiple timepoints. The proportion of patients achieving HAM-D remission was higher for both brexanolone groups compared with placebo, with the BRX60 group achieving statistical significance at multiple timepoints, including the 60 h primary endpoint [51% of patients in the BRX60 group versus 16% in the placebo group; OR 6.0 (95% CI 2.1–17.8), p = 0.0011]. Findings on the CGI-I mirrored that of the HAM-D, with both BRX90 [82% of patients, OR 4.0 (95% CI 1.4–11.6), p = 0.0095] and BRX60 [84% of patients; OR 4.0 (95% CI 1.3–11.7) p = 0.0131] achieving a higher proportion of patients improving relative to placebo at 60 h (56% of patients).
In study two, 108 patients were randomized to receive BRX90 (n = 54) or placebo (n = 54). On the primary endpoint, the mean reduction in HAM-D total score from baseline at the end of the 60 h infusion was 14.6 points (SE 0.8) in the BRX90 group compared with 12.1 points (SE 0.8) in the placebo group [mean difference −2.5 (95% CI −4.5 to −0.5), p = 0.0160]. The decrease in HAM-D total score for the BRX90 group relative to placebo reached statistical significance at hour 48 and remained significant at subsequent timepoints (60 h, 72 h, and 7 days). Although the BRX90 group retained its decrease in HAM-D total score magnitude at 30 days [−14.6 (SE 0.8) at 60 h versus −14.7 (SE 1.0) at 30 days], there was a marked improvement in placebo HAM-D total score at that timepoint [−12.1 (SE 0.8) at 60 h versus −15.2 (SE 0.0) at 30 days] resulting in BRX90 not being statistically or numerically superior. The proportion of patients achieving HAM-D symptom response in the BRX90 group compared with placebo reached statistical significance at all timepoints between 24 h and day 7. The proportion achieving HAM-D remission was also higher in the BRX90 group than placebo at all timepoints between 8 h and day 7, achieving statistical significance at multiple timepoints. At hour 60, coinciding with the conclusion of brexanolone infusion, 61% of patients in the BRX90 group versus 38% of patients in the placebo group achieved HAM-D remission [OR 3.4 (95% CI 1.5–7.9), p = 0.0033]. Similar to study one, findings on the CGI-I mirrored that of the HAM-D. At 60 h, the proportion of patients achieving CGI-I response was significant higher in the BRX90 group (79.6%) than the placebo group [55.8%, OR 5.0 (95% CI 2.0–12.5), p = 0.0005], and the results were sustained at day 7.
Tolerability
In the open-label proof-of-concept trial, adverse events (AEs) were reported by all four patients. All AEs were mild or moderate in severity, self-limited, and no patients discontinued brexanolone because of an AE. Sedation was the most common AE and was reported by two patients. 48 In the phase II trial, the tolerability of brexanolone was assessed by recording AEs, laboratory measurements, vital signs, and electrocardiograms (ECGs). 48 Sedation and emergent suicidal ideations were assessed utilizing the Stanford Sleepiness Scale and Columbia-Suicide Severity Rating Scale (C-SSRS), respectively.54,55 AEs were more common in the placebo group (8 of 11) than the brexanolone group (4 of 10), and there were no serious AEs, deaths, or discontinuations in either group. The most frequent AEs in the brexanolone group were dizziness and somnolence (each occurring in 2 of 10). Moderate AEs in the brexanolone group consisted of one patient developing sinus tachycardia and one patient developing somnolence. There were also two moderate AEs in the placebo group; one person developing infusion-site pain, and another developing a tension headache. There were no differences between groups on the Stanford Sleepiness Scale before or after treatment. Suicidal ideations improved in both groups, and no individuals experienced a worsening of suicidal ideations during treatment or follow up.
Brexanolone was well tolerated in both phase III studies, and safety and tolerability assessment measures were consistent with the phase II trial (Table 1). 29 Adverse events occurring greater than 5% for drug and at least twice the rate of placebo include: (a) somnolence, occurring at 6% in the placebo group, and 21% and 13% in the BRX60 and BRX90 groups, respectively; (b) flushing/hot flush occurring at 0% in the placebo group and 5% in the BRX60 group; and (c) dry mouth, occurring in 1% of the placebo group and 11% of the BRX60 group.
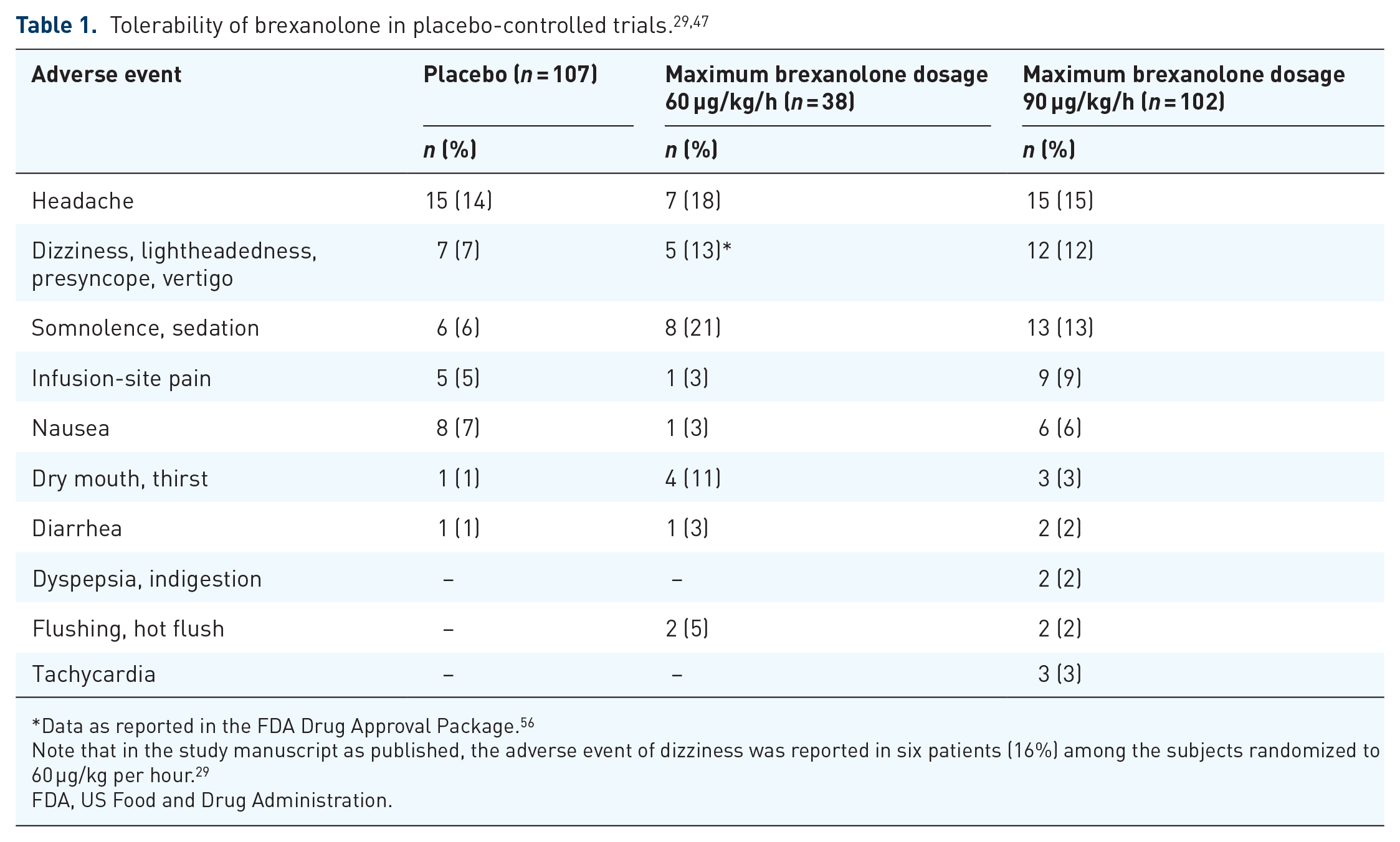
Data as reported in the FDA Drug Approval Package. 56
Note that in the study manuscript as published, the adverse event of dizziness was reported in six patients (16%) among the subjects randomized to 60 µg/kg per hour. 29
FDA, US Food and Drug Administration.
Across study one and two, five (4%) patients had excessive sedation considered to be due to brexanolone. All sedation events were completely resolved within 90 min with no respiratory or hemodynamic sequelae. However, based on the AEs which included dizziness, sedation, and loss of consciousness, brexanolone was approved for use through a Risk Evaluation and Mitigation Program (REMS), and a ‘black box’ warning was included. 57 The ‘black box’ warning states that patients are at risk of excessive sedation and sudden loss of consciousness during the administration of brexanolone. Brexanolone is a Schedule IV controlled substance with a theoretical abuse potential. In a human abuse potential study, 3% of subjects administered brexanolone 90 µg/kg (maximum recommended infusion rate) and 13% administered brexanolone 270 µg/kg (three times maximum recommended infusion rate) reported euphoric mood, compared with none administered placebo. 50
The effect of brexanolone on the QT interval was evaluated in 30 healthy adult subjects in a phase I randomized, placebo- and positive-controlled, double-blind, crossover study. At dosages 1.9 times higher the highest recommended infusion rate (90 µg/kg per hour), brexanolone did not prolong the QT interval to a clinically relevant extent. 50 The impact of brexanolone on lactation was studied in an open-label, phase Ib trial, involving 12 healthy, lactating women, who were ⩽6 months postpartum. 58 Each participant received a 60 h brexanolone infusion titrated to the maximum of 90 ug/kg per hour. Expressed breastmilk was not given to infants during the study. Breastmilk and plasma concentrations were measured and found to correlate. The concentration of allopregnanolone was less than 10 ng/ml in more than 95% of women by 36 h after the infusion, and below the limit of detection (5 ng/ml) in the majority of women by 72 h. There was no apparent accumulation of allopregnanolone in plasma or breastmilk. 58 The relative infant dose [RID (an estimate of infant drug exposure via breast milk)] was 1–2%. 50 An RID ⩽10% is associated with decreased risk. 59 Given such a low RID, it is most likely safe to breastfeed during pregnancy; however, pumping and disposing of breastmilk until after the infusion is a reasonable option as well.
Barriers to administration
Brexanolone is a complex intervention to deliver and entails substantially more obstacles than a prescription for an oral antidepressant medication. Based on the ‘black box’ warning regarding excessive sedation or sudden loss of consciousness during administration, a mandated REMS must be adhered to. Patients are required to stay at the infusion center/hospital for the duration of the 60 h treatment. The safety and monitoring requirements will likely preclude brexanolone from being administered on an inpatient psychiatric unit where infusions are not ordinarily administered, making a medical floor, obstetrics and gynecology floor, or 24 h infusion center a more likely and appropriate setting. A hospital setting would allow for easier monitoring by both psychiatric and obstetric clinicians.
A healthcare provider must be available onsite to continuously monitor the patient and intervene as necessary for the duration of the infusion. Pulse oximetry equipped with an alarm is necessary; should pulse oximetry reveal hypoxia, the infusion should be immediately stopped (and not resumed). Patients need to be assessed for excessive sedation every 2 h during planned, non-sleep periods. Initiation is recommended for early enough in the day to allow for recognition of excessive sedation. In clinical studies, 4% of brexanolone-treated patients compared with 0% of placebo-treated patients experienced a loss of consciousness that required dose interruption or reduction.29,47 A higher percentage of brexanolone-treated patients who used concomitant antidepressants reported sedation-related events. 50 In the event of a loss of consciousness or excessive sedation, the infusion should be held until the adverse event has resolved, and then possibly restarted at the same or lower dose.
As a consequence of these barriers, few physicians will have had experience using brexanolone. This lack of firsthand knowledge can be an impediment to brexanolone prescribing. Knowledge of brexanolone is relevant for clinicians working with women of childbearing age and includes primary care, psychiatric specialty care, and obstetric specialty care. A referral network may help improve access to infusion treatment with brexanolone.
Lastly, a major barrier remains the under-recognition of PPD. There are many possible reasons for the under-reporting of PPD, one of which includes patient reticence to receive pharmacologic treatment. In a study of 509 well-educated, high-income women, only 33% felt it was acceptable to take psychotropic medications during pregnancy, and only 35% when breastfeeding. 60 Women listed the most common barriers to receiving treatment for depression as ‘no time’ (64.7%) and ‘stigma’ (42.5%).
Cost
The financial impact of PPD can be substantial, and attributable to a multitude of factors, including, but not limited to: postpartum mother and child morbidity and mortality; lost productivity due to depression; and increased hospital costs. 61 A potential limitation of brexanolone is the estimated cost of $34,000 for one continuous infusion, excluding hospitalization costs, whereas selective serotonin reuptake inhibitors cost a very small fraction of this price and are often successful in treating PPD.62,63 This may preclude many uninsured and insured patients from receiving brexanolone. 64 In clinical practice, patients receiving brexanolone are often already receiving treatment with oral antidepressants. In the brexanolone clinical trial program, 22–36% of patients were receiving concomitant antidepressant medication, with no impact on brexanolone effectiveness. 28 Complicating this matter further is the lack of uniformity across differing healthcare insurers. Commercial and government insurance coverage varies by provider, with each insurer having different requirements for treatment coverage. Insurance companies can differ in the diagnostic criteria for PPD that they will accept [Diagnostic and Statistical Manual of Mental Disorders, 5th edition (DSM-5) criteria versus diagnostic criteria used in brexanolone clinical trials] and the clinician diagnosing PPD (psychiatrist versus obstetrician). In addition, it is not uniform what the insurer will cover, with some covering medication costs and others covering the cost of hospitalization. This is analogous to the insurance coverage for transcranial magnetic stimulation, in which, especially after it was first FDA approved, there was a lack of uniform acceptance and criteria.
Discussion
PPD impacts approximately 10–20% of postpartum women, though the real number is likely higher due to under-reporting.9,10 Impacted mothers have increased rates of morbidity and mortality, including lost productivity and suicide. 16 The impact extends beyond the mother, affecting the mother–child dynamic and continuing to impact the child throughout adolescence with a delay in height, weight, emotional and behavioral development, and academic performance.17 –19 Evidence supports a multifactorial pathophysiology of PPD, with contributors ranging from environmental, to genetic, to hormonal, to neurotransmitter dysfunction, as well as interactions between these factors. Although the etiology of PPD is unclear, hormonal changes during pregnancy and postpartum are felt to play an important role for some women. A possible mechanism centers around allopregnanolone, an endogenous progesterone metabolite which is believed to act on the GABA-A receptor. 30 Allopregnanolone levels rise throughout pregnancy before falling precipitously following childbirth. Failure of the GABA-A receptors to adapt to falling allopregnanolone levels may precipitate PPD.35 –37
Allopregnanolone has poor oral bioavailability. Brexanolone is a proprietary formulation of allopregnanolone that can be administered intravenously to produce a stable serum level, thus potentially mitigating the precipitous decline that occurs following childbirth. 47 It has a rapid onset of action that is critical in alleviating symptoms at this important time in the life of the mother and baby. Mechanistically, it may be superior to traditional antidepressant options in some women, as it is designed to treat perinatal depression, rather than an episode of MDD that occurs during the perinatal period. The efficacy of brexanolone has been established in a total of three phase II/III studies. In each study, based on the change from baseline on the HAM-D rating scale, brexanolone was found to be more efficacious than placebo at 60 h, coinciding with the end of the brexanolone infusion. The improved HAM-D score was maintained at day 30, and CGI-I scores mirrored that of the HAM-D. In addition, brexanolone resulted in higher rates of remission than placebo at multiple timepoints. However, a limitation is the absence of any identified longer-term efficacy data.
In addition to being efficacious, brexanolone was fairly well tolerated, and there were no unexpected AEs or deaths. 29 When combining the phase II and III trials, 50% (n = 70/140) complained of any AE, and 0.036% (n = 5/140) suffered a serious or severe AE in the brexanolone groups, compared with 50.5% (n = 54/107) with any AE, and 0.019% (n = 2/107) with any serious or severe AE in the placebo groups.29,47 At this time, although breastfeeding while receiving brexanolone is not recommended due to a lack of information on the effect, the risk is likely very low and can be safely resumed 36–72 h following the completion of brexanolone therapy.
There are several accessibility concerns with brexanolone. Cost will likely be prohibitive in many circumstances, creating a significant barrier. Because of the risk of side effects, brexanolone needs to be administered under monitoring in a healthcare facility for the duration of the 60 h infusion based on its approval through a REMS program. Brexanolone is unlike any medication currently available for the treatment of depression or PPD, and as such, physicians have limited experience utilizing the medication, potentially making them less likely to recommend it. Limited prescriber experience has been shown as a barrier to prescribing other medications. 65
Bridging the gap
The barriers to identifying and treating PPD across specialties are multifaceted and far-reaching. A comprehensive review of these gaps is beyond the scope of this review and has been explored elsewhere. 66 One gap is the inadequate communication between specialties, and the existence of the silo model of healthcare which limits communication. Although effective communication between medical specialties is necessary for optimal patient care, it has been repeatedly found lacking. 67 One factor is a lack of uniform definitions and guidelines. Similar to how different specialties may define the same illness presentation as being either delirium or encephalopathy, PPD is often approached differently by psychiatry and obstetrics. The APA and ACOG have published differing guidelines and definitions for PPD, with a lack of common language, which can create diagnostic and treatment confusion. For example, ACOG includes the term ‘minor depression’ in their definition of PPD, which is a term not mentioned in the DSM-5. Moreover, screening tests used in obstetric practice (such as the EPDS) may be unfamiliar to psychiatric clinicians. Open and direct communication between specialties and being mindful of differing definitions, screening tests, and treatment approaches can help optimize patient care and reduce confusion.68,69 The psychiatry consultation service is often referred to as a consultation liaison (CL) service. By taking this liaison title to heart, interdisciplinary care will benefit. In the future, interdisciplinary guidelines and collaborative care between psychiatry and obstetrics would be an area of shared benefit. 70
Future considerations: sage-217
Brexanolone is only available as an intravenously administered formulation, limiting its utility as an easily administered treatment for PPD. However, an oral drug is being developed, zuranolone (sage-217), which shares a similar molecular pharmacologic profile with brexanolone. A complete discussion of zuranolone is beyond the scope of this review; however, given its investigational nature and close relationship to brexanolone, it warrants a brief mention. Zuranolone is an investigational, orally bioavailable positive allosteric modulator of synaptic and extrasynaptic GABA-A receptors. 71 It has a neuroactive steroid backbone which has been chemically modified to increase oral bioavailability and is dosed at bedtime. The terminal phase half-life of zuranolone is 16–23 h, compared with brexanolone which is approximately 9 h.50,72 Whereas brexanolone is chemically identical to allopregnanolone, zuranolone is not. In a multicenter, randomized, double-blind, parallel-group, placebo-controlled study, the efficacy and tolerability of zuranolone was studied in ambulatory adult women diagnosed with severe PPD. A total of 151 women were randomized (1:1) to receive zuranolone (30 mg once daily) or placebo for 2 weeks. The primary endpoint was the change from baseline in the HAM-D total score at the end of treatment (day 15). Individuals treated with zuranolone had significantly greater improvement in HAM-D total score at day 15 versus placebo (change from baseline −17.8 versus −13.6 respectively, p = 0.0028). 73 Significant differences occurred as early as day 3 and were sustained through day 45. On secondary endpoints, HAM-D response at day 15 was 72% versus 48% in patients treated with zuranolone versus placebo (p = 0.005), and remission was 45% versus 23% (p = 0.011). 74 MADRS and CGI-I results mirrored that of the HAM-D. 75 Zuranolone was generally well tolerated, and the AEs in the zuranolone group were 60.3% versus 52.1% in the placebo group. The number of patients with serious or severe AEs were consistent in each group, and there was no increase in suicidal ideation in either group, as measured by the C-SSRS. 73
Zuranolone is also being studied for MDD. Rationale for use includes the observation that decreased levels of allopregnanolone have been detected in the cerebrospinal fluid of individuals suffering from MDD, normalizing after treatment with antidepressants, and on studies that support the GABAergic pathophysiology of MDD.76,77 In a randomized, double-blind, placebo-controlled phase II trial of zuranolone in patients with MDD, zuranolone was found having superior efficacy when compared with placebo on its primary endpoint. 71 In the study, 89 patients were randomized in a 1:1 ratio to receive zuranolone or placebo. Patients were men and women, and 18–65 years of age. However, in a larger pivotal phase III trial, zuranolone failed to meet its primary endpoint of change from baseline to day 15 on the HAM-D when compared with placebo. 78
Conclusion
Brexanolone, chemically identical to the neuroactive steroid allopregnanolone, represents a novel option for PPD and is the first FDA approved treatment for this indication. When compared with existing treatment options, brexanolone represents a unique mechanism of action, functioning by acting on the GABA-A receptor as a positive allosteric modulator. GABAergic signaling has been implicated in the etiology of depression, and neuroactive steroids, which are potent GABA modulators, may play a role. Given the cost of brexanolone, treatment is expected to be reserved for patients with severe PPD, and in many circumstances, there may be logistical and practical barriers that prevent the initiation of brexanolone treatment. In the future, comparison studies between brexanolone and conventional antidepressants, and reporting of longer-term outcomes following brexanolone treatment, would be beneficial.
Footnotes
Conflict of interest statement
Justin Faden, DO: No conflicts of interest.
Leslie Citrome MD, MPH: In the past 12 months, consultant: AbbVie, Acadia, Alkermes, Allergan, Avanir, BioXcel, Cadent Therapeutics, Eisai, Impel, Intra-Cellular Therapies, Janssen, Lundbeck, Luye, Merck, Neurocrine, Noven, Osmotica, Otsuka, Sage, Shire, Sunovion, Takeda, Teva. In the past 12 months, speaker: AbbVie, Acadia, Alkermes, Allergan, Eisai, Intra-Cellular Therapies, Janssen, Lundbeck, Merck, Neurocrine, Noven, Otsuka, Sage, Shire, Sunovion, Takeda, Teva. Stocks (small number of shares of common stock): Bristol Myers Squibb, Eli Lilly, J & J, Merck, Pfizer purchased >10 years ago. Royalties: Wiley (Editor-in-Chief, International Journal of Clinical Practice, through end 2019), UpToDate (reviewer), Springer Healthcare (book), Elsevier (Topic Editor, Psychiatry, Clinical Therapeutics).
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.