
Abstract
Multiple clinical trials have shown that aspirin can reduce all cardiovascular events in primary and secondary prevention and yet there is a large population in whom aspirin fails. This review brings together the evidence and controversies surrounding the definition of ‘aspirin treatment failure’, its clinical significance and the possible approaches to managing such patients. Several different assays have been developed to measure the biochemical action of aspirin. At present there is no ‘gold standard’ and there is massive disparity between methods. Studies thus far have shown inconsistent results and to date the treatment of aspirin therapy failure is left to the discretion of the leading physician.
DECLARATIONS
None declared
None declared Ethical approval Ethical approval was not applicable
FS
FS is the original author of this review paper. MA has acted as senior author to this review article
Hesketh Roberts
Introduction
Aspirin is one of the most widely prescribed pharmacological agents. Since discovery in 1967 much has been elucidated about its antithrombotic effects. 1 Multiple clinical trials have shown that aspirin can reduce all cardiovascular (CV) events in primary and secondary prevention. The Antithrombotic Trialist Collaboration, which is the largest meta-analysis to date, analysing 287 randomized trials, found the absolute risk reduction to be in the region of 32%. 2
Aspirin achieves its effect by irreversibly inhibiting the cyclo-oxygenase (COX) enzymes, thus preventing the conversion of arachiodonic acid to one of many eicosanoids, which include thormboaxane. This halts the vasoconstrictive and platelet agonist effect that would otherwise take place. This antagonistic effect only lasts for 10 days, which is the life span of platelets. 1
Despite its common use, it is thought up to 75% of patients may not benefit from this risk reduction. 1 This was shown by a recent study on platelet aggregation in which up to 20% of serious vascular events in high-risk patients may have been attributable to failure of aspirin to exert a therapeutic effect. 3 This phenomenon has been termed ‘aspirin treatment failure’ and various clinical and laboratory methods have been developed to investigate it. Furthermore, the ASCET study found that aspirin treatment failure was likely a true phenomenon but also very much assay dependent. 4 This study highlighted the fact that aspirin treatment failure in the form of platelet inhibition was found to be much more striking when Arachodonic acid was not the used stimulus. 5 This raised important issues. Firstly, what is the gold standard for measuring aspirin treatment failure? Secondly, that this very real phenomenon requires further insight as the consequences of cardiovascular morbidity may be deemed significant.
Arguably the most compelling aspect to the existence of aspirin treatment failure is the most recent research in 2012. A study involving more than 1000 patients all on single treatment aspirin were randomized to continue aspirin or switch to Clopidogrel. Using a platelet function analyser this study found that patients on aspirin did have a higher predisposition if having suboptimal platelet levels or a deficiency in Von Willebrand factor. 5 This brings to light the definition of aspirin treatment failure which can be applied to any situation in which a patient experiences cardiovascular endpoints despite treatment dose aspirin. The aetiology of such a cause can be multi factorial, as will be explored further in this review.
This review explores the evidence and controversies surrounding the definition of aspirin treatment failure, its aetiology, its clinical significance and the possible approaches to managing patients with ‘aspirin treatment failure’ (ATF).
Evidence used in this review article was gathered using literature research. Both primary evidence and research articles have been studied and information and evidence has been compiled from these to provide the reader with a concise and up to date review of aspirin treatment failure. The diagrams and tables used have been independently formulated.
Diagnostic methods
At present there is no gold standard for identifying ATF. Methods include light transmission aggregometry platelet function analysis and assays measuring thromboxane metabolites (Table 1 provides a summary of the various methods used to detect ATF along with their advantages and disadvantages). The difficulty in making a laboratory diagnosis of ATF is limited by individual patient complexity and lack of correlation with each test. For example, a patient having multiple CV events despite being on treatment dose aspirin would suggest ATF. Utilizing one assay may show normal measurements whereas another assay may be abnormal. Reproducibility and robustness remain weak with all assays, and they serve only as surrogate markers.6–8
Summary of the various methods of investigating Aspirin Treatment Failure (compiled independently by the leading author)

Patients who undergo recurrent ischaemic events despite the correct therapeutic dose of aspirin warrant biochemical investigation. As stated, no gold standard method exists. Although LTA is the most reliable, economically its use is not yet widely available. Serum thromboxane reflects the total capacity of platelets to synthesize TXA2. This is arguably the most specific test as contribution by other blood cells in this process is marginal. At present this is likely to be the test of greatest viability.
Notably, studies have shown that regardless of the test used a total lack of response with regards to platelet responsiveness is rarely zero. The variety in test results is dependent on the specificity of the method used, as described above, with percentage response levels ranging from 5–61%. 7
Causes of biochemical ATF
The most intensely studied areas include:
increased platelet sensitivity to collagen;
platelet activation by pathways not suppressed by aspirin;
thromboxane synthesis through pathways not inhibited by aspirin. 9
Increased platelet sensitivity to collagen
Injury to the vascular endothelium exposes underlying collagen, which can act as an adherence site for platelet aggregation. In vitro studies using single-base mutation platelets have shown this affinity for collagen to be heightened. When therapeutic doses of aspirin are used to counteract this, they are ineffective. 9 A case-controlled study on eight healthy men in Japan with such mutations of collagen found that three of the individuals showed signs of aspirin resistance, determined by shorter than expected bleeding time and positive platelet aggregation assays. Furthermore, synthetic concentrations of collagen that were required to produce a 90% platelet aggregation were 5% higher in the aspirin non-responders due to collagen mutations. These results were reproduced when the trial was repeated. 10
Thromboxane synthesis by pathways not suppressed by aspirin
More recent studies have found that variants in COX-1 and COX-2 levels or polymorphisms contribute to ATF, with COX-2 having a greater effect when patients are on therapeutic doses of aspirin. 11 Both these enzymes are involved in the synthesis of thromboxane. Aspirin works by reducing thromboxane production in platelets. The dosing, however, is vital to aspirin's action because, in addition to the function mentioned above, higher doses of aspirin inhibit prostacyclin (PGI2). PGI2 is an inhibitor of platelet aggregation at the endothelial level. Research has found that the doses used in clinical practice are adequate to prevent platelet aggregation without reducing the levels of PGI2. This would conversely cause platelet aggregation (Figure 1).
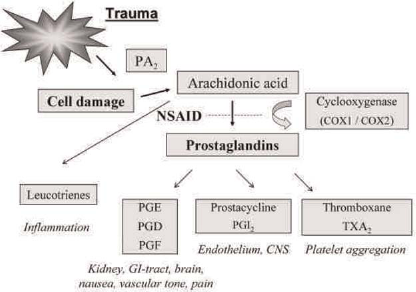
Mechanism of action of aspirin at the site of cyclo-oxygenase enzyme activity
Aspirin blocks COX-1 enzyme function as it covalently binds to and acetylates the serine hydroxyl group in its hydrophobic pocket. This inhibits the passage of arachidonic acid to the so-called active site of the enzyme. However, despite aspirin being commonly referred to as ‘a non-selective COX inhibitor’, its affinity for COX-1 is 50–100 times greater than COX-2. 12
Aspirin at serine 516 covalently acetylates COX-2. However this does not stop the enzyme from oxidizing arachidonic acid, providing an alternative pathway of platelet aggregation. 13 Although there is 90% homology in structure between COX-1 and -2, it is the 10% variability that seems to allow COX-2 to escape inhibition and exert a pathway for thromboxane production.
A retrospective study carried out on patients in Germany who had undergone coronary artery bypass grafting found that those who failed to show decreased thromboxane production had immune-assay levels of COX-2 higher than that of the control group. 14 However, studies to the contrary have been cited earlier. 15
The role of genetic polymorphisms in relation to the COX family has also been studied. 16 There have been reports that single-nucleotide polymorphisms (SNPs) in COX-1 and -2 are responsible for variability in aspirin effect. However, such findings seem to be isolated events, and in fact, certain polymorphisms have shown an increased sensitivity to aspirin. 17
The role of COX-2 polymorphism, coined COX-4, holds more promise. 16 A case-controlled study on 1700 Italian patients showed that an SNP led to a COX-2 enzyme with lower plaque formation in patients post Myocardial Infarction (MI) and also questions whether the lower incidence of MI and stroke in northern Europe compared with the Mediterranean is entangled in the genetic heterogeneity between these populations and not solely due to lifestyle factors. 16
Platelet activation by pathways not suppressed by aspirin
Glycoprotein IIb/IIIa is a platelet membrane receptor, which functions to bind to fibrinogen and von Willebrand factor. The GPIIIa polymorphism can influence both platelet activation and aggregation by increasing the cell surface expression of the receptor and increasing its affinity for fibrinogen, hence increasing the efficacy of platelet aggregation. 18 Alleles of this receptor, termed polylactic acid (PLA1 and 2) have both been associated with altered platelet binding and thus prolonged bleeding times. 19
A recent study looked at 20 patients with stable coronary heart disease in whom aggregation methods showed those with PLA2 genotype to have a reduced sensitivity to aspirin. 20 In contrast, alternative polymorphisms have reduced platelet function. Interestingly, a clinical study carried out on patients having undergone coronary artery stenting showed no significant difference between those with the PLA1/2 genotype and control. 21 However, the endpoint to this study differed from previous studies and was based on symptomatic patients. This polymorphism is the most extensively studied in relation to ATF and thus far 19 studies encompassing 1389 patients have been carried out. 21 However, the cohort population in each study was small and there has been a significant heterogeneity shown in these studies that warrants further research into this promising area.
In addition, conditions such as diabetes, dyslipidaemia, smoking and heart failure cause the production of non-platelet-derived sources of TXA2. This can increase the lipid peroxidation of arachidonic acid, serving as the precursor for thromboxane production.22, 23, 23
Causes of clinical ATF
Compliance
In many cases non-compliance with aspirin therapy is the leading cause of failed therapeutic effect. Ten percent of patients on aspirin for secondary prevention were found to be non-compliant with their medication. 25 In reality this is likely to be even higher. 26
Dosing
Appropriate aspirin dosing should also be considered in patients complying with medication. Biochemical assay research shows that there is a correlation between aspirin dosage and platelet inhibition. Studies have shown that the anatomical position of the thrombus in the cardiovascular system has an effect on whether low dose aspirin will be of benefit. In patients with a cardiovascular event doses of aspirin between 75 and 100 mg daily have been shown to be efficacious. Doses beyond this range are no more efficacious as COX receptors are fully saturated. 27
In an acute stroke aspirin doses of 300 mg are sufficient to inhibit TXA2 production. Lower doses can also achieve this effect but can take up to 10 days to exert the same inhibitory effect as higher dose aspirin. 28
Interactions
Drug interactions between non-steroidal anti-inflammatory drugs (NSAIDs) and aspirin can lower aspirin's therapeutic effect. As with aspirin, these drugs also inhibit COX-1, but unlike aspirin they are reversible inhibitors of COX. NSAIDs compete with aspirin for the active site on COX-1 and may reduce its antithrombotic effect. 29 Use of selective COX-2 inhibitors does not pose such an obstacle. 30 However, several studies have suggested selective COX-2 inhibitors increase the probability of ischaemic cardiac events although the reason remains unknown (Table 2 summarises the likely causes of ATF and their aetiology).31, 32
Summary of the likely causes of aspirin resistance and their likely aetiology

ATF, aspirin treatment failure; PLA1 and 2, polylactic acid1 and 2; NSAIDs, non-steroidal anti-inflammatory drug; COX-1/2, cyclo-oxygenase 1/2
Clinical significance of ATF
Snoep et al. 7 carried out a meta-analysis on 1813 patients across 12 studies. Biochemical ATF was found in 27% of patients and when compared with a cardiovascular endpoint, these patients had a statistically more significant adverse outcome. Furthermore, a meta-analysis of 6450 patients across 64 different populations found that those patients deemed aspirin non-responders using PFA-100 were at a significantly higher risk of vascular events than healthy subjects sensitive to aspirin. 7
Numerous studies have shown a similar pattern of ATF patients being at higher risk of serious vascular events. However, currently insufficient trials are available in which the cohort of ATF subjects closely mirrors the control group in relation to the power of the study and also co-morbidities.
In a more recent study, however, more than 1000 patients who had previously had a cerebrovascular event were retrospectively analysed to determine whether patients were at increased risk of recurrent stroke or death while on aspirin. A three-month and one-year follow-up concluded that those patients deemed ATF were at no higher risk of recurrent events than those deemed aspirin responsive. 33 Even though one cannot decipher if at a molecular level (as previously discussed) these patients were homogeneous in their responsiveness to aspirin, it does ask the question as to whether ATF is indeed of clinical significance.
Violi et al. in 2006 emphasized that aspirin treatment failure can only be said at present to be a subjective phenomenon and not one to be universally accepted. Due to the great patient variances with regard to co-morbidities and the multifactorial aspects of atherosclerosis, a cause-effect relationship cannot be directly applied. 34 This point is further elaborated by the fact that laboratory values of platelet activation have been reported in the ranges of 21–78%. Depending on the method and accuracy of measuring platelet activation some trials have found no evidence of ATE.35, 36
Thus one can argue that the term ATF cannot be formally acknowledged until there is a gold standard by which our trial methods are conducted and reference ranges agreed upon. Additionally with many of these studies there is a lack of clinically defined endpoints and the issue of confounding variables.
However, what cannot be ignored is the fact that patients deemed non-responsive to aspirin therapy are at higher risk of mortality. It is for these select individuals that measures to combat aspirin failure need to be found.
Measures to combat ATF
In order to identify a patient as having ATF a clear definition must first be sought. In clinical terms it is the occurrence of ischaemic events despite treatment optimisation. As already discussed, forming a biochemical definition is problematic, as no gold standard method of testing exists. However, in its broadest terms it would be at the point where aspirin is no longer able to inhibit COX-1-dependent TXA production.
With this definition in mind steps must be put in place to reduce the incidence of recurrent ischaemic events in such patients. The following treatment options are available to the clinician, some of which are well established, others less so, and require risk-benefit stratification at an individual patient level.
Treatment of individual risk factors
The most logical and arguably safe method of treating ATF is by identifying and treating non-atherosclerotic risk factors of the qualifying vascular event that may not respond to aspirin in the first instance. For example use of steroids for arteritis causing stroke. One should ensure that compliance issues are addressed with the patient and patient education is at the forefront of addressing such issues. Ensuring that the patient's medications are reviewed on a regular basis is vital. Drug interaction as previously mentioned is an area of drug resistance that should not be underestimated. For example, ensuring the patient is not taking NSAIDs along with aspirin is a simple but effective measure of dealing with a possible cause of aspirin treatment failure.
Increasing the dose of aspirin
Doses of aspirin as low as 30 mg/day have been shown to inhibit platelet aggregation in up to 40% of subjects. 1 A further 10% of subjects respond to doses of 100 mg of aspirin. In the remaining population doses of 300–500 mg of aspirin cause complete platelet inhibition. However, on a clinical level doses as high as 300 mg are only used in the setting of an acute cerebrovascular incident and doses as high as this for a persistent length of time may not justify the adverse events that accompany such high doses.
Other studies have shown inconsistent results but with a smaller study population and doses of aspirin not exceeding 100 mg once a day. 2 This may be attributable to genetic polymorphisms and would suggest ATF. On an individual basis patients who have suffered from cardiac or cerebrovascular events do not seem to show any benefit from aspirin doses above 100 mg. Recent studies attribute this to the multifactorial disease profile of patients, i.e diabetes, obesity, smoking. Whether or not such hypothesis holds true is the next challenge in the aspirin scenario. For this present time the clinician should look to optimise aspirin dose within this range and then look to alternative pharmacological intervention, as mentioned below. 37
Clopidogrel
This drug is part of a group termed thienopyridines. Clopidogrel works by inhibiting the action of the ADP receptor and thus preventing the biochemical pathway that allows for the activation of the glycoprotein IIb/IIIa receptor.
Pamukcu et al. showed that out of 52 patients that failed to benefit from aspirin therapy, in a cohort of 234, the 50% of which were on dual therapy showed a marked reduction in cardiovascular events after a 12-month follow up. Additionally, 28 of the patients enrolled that did respond to aspirin therapy were split into two groups, those in whom Clopidogrel was continued and those in which it was prematurely stopped. Those on dual therapy had significantly fewer cardiovascular events. 38 However, the study sample was small. It must be taken into account that approximately 50% of patients that are aspirin resistant are also Clopidogrel resistant.
The CAPRIE study further showed that Clopidogrel is deemed as having equal efficacy to aspirin in relation to incidence of stroke and MI. 39 The aetiology behind such a phenomenon is several fold. One theory is that the CYP3A4 gene is mutated in subjects resistant to Clopidogrel and thus it cannot be converted to its active metabolite. Other intrinsic mechanisms include PY2 receptor variability, increased release of adenine dinucleotide phosphate or up regulation of other platelet regulator pathways. Further work has shown an even larger impact on high-risk populations such as those with previous coronary artery bypass grafts, dyslipidaemia or multiple vascular beds.
The most significant finding is arguably that of the CURE study that showed the use of Clopidogrel with aspirin in both the acute coronary syndrome and percutaneous coronary intervention setting produced a 30% risk reduction in endpoints. Clopidogrel is a well-established dual antiplatelet therapy for patients postcardiological intervention. 40
The clinician must therefore ensure that Clopidogrel is initiated in patients with recurrent cerebrovascular events and also in the setting of interventional cardiological procedures. The argument of the optimal dosing of Clopidogrel is ongoing, with some studies showing higher dosing of Clopidogrel may be beneficial. However, this is yet to be internationally agreed, apart from in the acute setting. 41
Therefore in patients in need of dual antiplatelet therapy Clopidogrel is a well-established first-line agent. The hypothesis of Clopidogrel resistance is very much an unknown quantity and as yet not something clinically one should consider as research is lacking in this field. As a side note the use of dipyridamole in secondary prevention in stroke is no longer warranted. Studies comparing aspirin and dipyridamole to use of Clopidogrel showed no significant superiority to the former use. Therefore, recommendations are now for the use of Clopidogrel rather than dipyridamole in patients with high risk or recurrent cerebrovascular incidents. 41
Prasugrel
In the UK prasugrel has been granted use in combination with aspirin in those who have had stent thrombosis during treatment with Clopidogrel and in diabetics. This agent, which is a third generation thienopyridine, may be more efficacious than aspirin and Clopidogrel. In the future, prasugrel may be used in those with ATE 42 There are now clear NICE guidelines regarding the use of prasugrel, with emphasis of its use in acute cardiovascular settings. One must emphasize that optimal use of prasugrel is with aspirin. 43
Most recently the PLATO study has been used by NICE to highlight that ticagrelor plus aspirin is superior to Clopidogrel plus aspirin. This was a large-scale study and showed that primary endpoints and adverse side-effects were significantly fewer with ticagrelor than with Clopidogrel. Guidelines issued by NICE now state ticagrelor should be used over Clopidogrel in ACS, STEMI and NSTEMI. At present however this protocol has not been adopted throughout the UK and thus Clopidogrel use for the time being is still apparent.
Conclusion
Although literature on ATF is limited, there is evidence that this may be a significant clinical entity. Targeting this group may significantly reduce the number of thrombotic (fatal and non-fatal) events. The first step in combating ATF will be agreement upon a universal method of testing. At present LTA is used in the majority of studies though not accepted as the ‘gold standard’.
The use of Clopidogrel, which is already established as an adjuvant to aspirin therapy, is a means by which people with biochemical ATF can have an improved prognosis. Even though data suggest that higher doses may not be beneficial, at present there is little alternative to this approach. In due course, however, it seems that ticagrelor will take over the role of Clopidogrel.
Due to the limited number of large randomized controlled trials in this field, labelling a patient as not responsive to aspirin may be unjustified. Co-morbidities and compliance must be considered as a primary reason why patients may continue to have cardiovascular events. However, the issue of ATF cannot be ignored as data are suggestive of this phenomenon.
A clear definition of ATF cannot be given until greater emphasis is placed on conducting trials on patients from similar cardiovascular backgrounds and in whom non-biochemical causes have been ruled out. Controversy remains as to whether ATF is a heterogeneous process or one of molecular origin. 44 Further work in this field is required in order to record the effects of reversing an apparent state of ATF with alternative treatment. Such methods may hold ethical issues, especially as limited data are available to conclude ATF as an entity is an indicator for increasing morbidity and mortality.
At present patients who have vascular events despite being on adequate antiplatelet therapy raise the suspicion of ATF. However, as the diagnostic methods are unclear, the decision to increase aspirin or add a second agent is left to the clinical discretion of the physician. Addressing ischaemic event risk factors and optimizing platelet therapy with established evidence-based research is at present the optimal therapy to offer our patients. The use of Warfarin has not been mentioned in this paper, solely as its use can be justified using risk evaluation parameters such as the CHADS2- VASC scoring system and is highly dependent on individual case of the patient.
Footnotes
Acknowledgements
None