
Abstract
Background:
Although there are reports of adverse events (AEs) of cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors, the safety of ribociclib alone has not yet been comprehensively evaluated in real-world clinical practice.
Objectives:
To investigate the overall real-world safety profile of ribociclib by mining data from the FDA Adverse Event Reporting System (FAERS).
Design:
A retrospective disproportionality analysis was conducted based on the FAERS database.
Methods:
We processed reports from the first quarter of 2017 to the second quarter of 2023 and applied disproportionality analysis using four different methods: reporting odds ratio, Medicines and Healthcare Products Regulatory Agency, Bayesian confidence propagation neural network, and multi-item gamma Poisson shrinker.
Results:
A total of 12,885 AE reports of ribociclib as the primary suspect were enrolled. 48.81% of AEs occur within 60 days of ribociclib administration. Blood and lymphatic system disorders and abnormalities in investigation at the system organ class level showed statistically significant signals in all four methods. Nausea (n = 1426), neutropenia (n = 940), vomiting (n = 863), white blood cell count decreased (n = 812), and alopecia (n = 536) turned out to be the five most frequent AEs at the preferred term level. Twenty-eight AEs undiscovered in the label were newly identified. Neutropenia, as a widely recognized AE, was observed to potentially result in more serious outcomes than previously anticipated (p < 0.001).
Conclusion:
This study utilized the FAERS database to analyze real-world AE signals associated with ribociclib following its market approval. We characterized the clinical profiles of reported AEs and found some significant signals consistent with previous clinical trials. In addition, several AEs not included in the drug label or exhibiting unexpected severity were detected. These findings provide valuable insights for clinicians and highlight directions for further causality-focused research to validate the observed results.
Plain language summary
Why was the study done? Ribociclib, as an anti-cancer drug mainly applied to breast cancer patients, is now widely used. However, its comprehensive safety profile remains unrevealed. The researchers aim to use the adverse event reports registered in the FDA Adverse Event Reporting System to give a general view of adverse events of ribociclib in the real-world clinical practice after its marketing. What did the researchers do? The researchers collected reports from the first quarter of 2017 to the second quarter of 2023, analyzed the data and detected the signals of adverse events associated with ribociclib at different levels utilizing four different statistical methods. How is the study designed? This study is a disproportionality analysis, which analyzed the past data from the FAERS database. What did the researchers find? The researchers analyzed 12,885 reports in which ribociclib was suspected as the primary cause of the adverse event. Here are the main findings: - Almost half of the adverse events occur in the first 60 days of ribociclib using. - “Blood and lymphatic system disorders” and abnormalities in “Investigation” showed significant signals in all four methods used. Nausea, neutropenia, vomiting, white blood cell count decreased, and alopecia were the five most common adverse events with ribociclib. These were consistent with previous clinical trials. - 28 adverse events undiscovered in the drug label were identified. - Among the widely recognized adverse events, neutropenia could result in more serious outcomes than common knowledge. What do the findings mean? This study could serve as a warning to clinicians and provide a direction for further studies. Follow-up researches based on long-term data or further prospective studies are necessary to validate the results of our study and to reveal the causality.
Introduction
Breast cancer has become the leading cause of cancer death in women. 1 Cyclin-dependent kinase 4 and 6 (CDK4/6) inhibitors, administrated in company with an aromatase inhibitor or fulvestrant, such as abemaciclib, palbociclib, and ribociclib, have significantly improved the prognosis for patients with endocrine therapy-resistant, advanced, or metastatic breast cancer with hormone receptor—positive and human epidermal growth factor receptor 2—negative, 2 by blocking Rb phosphorylation, halting the cell cycle in the G1 phase, and preventing tumor cell proliferation. 3
Ribociclib, approved by the US Food and Drug Administration (FDA) in 2017, is an orally available, selective CDK4/6 inhibitor. According to a report from Verified Market Research, the global ribociclib market size was valued at USD 6.2 billion in 2023, and is projected to reach USD 12.5 billion by 2030. 4 Compared to abemaciclib and palbociclib, ribociclib has a lower off-target rate and achieves higher maximum plasma concentrations (over 2 µg/ml) with a long half-life (exceeding 30 h). 5 Several large, randomized, double-blind, placebo-controlled phase III trials about ribociclib have reported positive results on prognosis.6–9
The most commonly observed adverse events (AEs) associated with ribociclib are hematological, gastrointestinal toxicities, and pain as claimed by the label. Among rare AEs, prolonged QT interval, which is concentration-dependent, is notably highlighted. Neutropenia, hepatobiliary toxicity, prolonged QT interval, and interstitial lung disease (ILD) or pneumonia are AEs emphasized as most likely to require discontinuation of ribociclib. In the series of MONALEESA trials, the incidence of Grade 3/4 neutropenia reached 52.1%, 57.1%, and 63.5% respectively, whereas the incidence of Grade 3/4 nausea was reported to be 2.7% in the MONALEESA-2 trial.6,7,9 Other key Grade 3/4 AEs worth special attention include hepatobiliary toxicity (14.4%, 13.7%, 11% respectively), prolonged QT interval (4.5%, 3.1%, 1.8% respectively), and ILD or pneumonitis (0.6%, 0.2%, 0% respectively). In a German multicenter trial, the most frequently observed serious adverse events (SAEs) were dyspnea (2.4%), pneumonia (2.0%), and nausea (1.8%). 10 In a recent case reported from Switzerland, a severe idiosyncratic hepatocellular pattern of drug-induced liver injury (DILI), classified as Grade 3 by the International DILI Expert Working Group, was linked to the use of ribociclib. 11 Of note is ribociclib associated with a higher rate of cardiac complications in comparison to palbociclib and abemaciclib. Sudden cardiac death has been observed in a patient with concurrent hypokalemia. 12
Although the safety of ribociclib has been primarily documented through the aforementioned clinical trials, these premarketing trials often have limitations, such as small sample sizes, specific selection criteria, and relatively short follow-up periods. Rare, unexpected, fatal AEs and SAEs might not be completely covered. Moreover, previous pharmacovigilance studies on CDK4/6 inhibitors focused more on the comparison between the three drugs13,14 instead of ribociclib alone or were limited to certain side effects, such as thromboembolism, 15 skin toxicities, 16 ILD, 17 or DRESS syndrome, 18 which only provided a partial view of real-world information about the drug.
This study aims to offer an all-around insight into the safety profile of ribociclib alone during its clinical application, based on the extensive individual case safety reports data from the FDA Adverse Event Reporting System (FAERS), a spontaneous reporting system with open access to information on AEs reported to the FDA by the pharmaceutical industry, healthcare providers, and consumers, mirroring the entire world.19,20
Methods
Data sources and data processing
All reports from 82 countries on 5 continents submitted to the FAERS from the first quarter of 2017 (FDA approval of ribociclib for marketing) to the second quarter of 2023 (the latest update when the study began), 26 quarters in total, downloaded from https://fis.fda.gov/extensions/FPD-QDE-FAERS/FPD-QDE-FAERS.html in the form of ASCII data packets have been imported into SAS 9.4 software (SAS Institute Inc., Cary, NC, USA) for data cleaning and analysis.
Based on the method recommended by the FDA concerning removing duplicate reports, we have selected the PRIMARYID, CASEID, and FDA_DT fields of the patient demographic and administrative information (DEMO) table, and then sorted them by CASEID, FDA_DT, and PRIMARYID in turn. First, for reports with the same CASEID, the one with the largest FDA_DT value has been retained. Second, for reports with the same CASEID and FDA_DT, the one with the largest PRIMARYID value has been kept. Notably, since the first quarter of 2019, a deleted report list has been included in each quarter’s data package. Therefore, after data deduplication, reports will be eliminated according to the CASEID in the deleted report list. In cases with accessible data where ribociclib, searched as “Kisqali,” “LEE011,” “Kryxana,” “Risarg,” “Valamor” or “Ribociclib,” is the primary suspect drug, we finally included detailed information, such as patient characteristics (gender as binary variable, age as continuous variable, reporting year as categorical variable), time to onset (TTO, continuous variable), and outcomes (binary variable).
The preferred terms (PTs) and the system organ classes (SOCs) in Version 26.1 of the International Medical Dictionary for Regulatory Activities (MedDRA®) dictionary have been obtained. However, several SOCs such as Injury, poisoning and procedural complications, Surgical and medical procedures, and Social circumstances were excluded as they were considered signals unrelated to the drug. Duplicate PTs have been deleted. A flowchart of the multistep process of our study is shown in Figure 1.

Flowchart of the multistep process including data extraction, processing, and analysis from the FAERS database.
Study design and statistical analysis
A 2D retrospective observational disproportionality analysis was conducted to determine the potential association between ribociclib and a certain AE, which obtained AE data for ribociclib from its combined use. Positive signal was defined when a given AE associated with ribociclib appeared at a significantly higher frequency than the background frequency of events caused by other reasons (e.g., other drugs and tumors) in the database, meeting a predefined threshold or standard.21,22
Disproportionality analysis was performed using SAS 9.4 software (Table 1). Statistical analysis methods utilized included reporting odds ratio (ROR), Medicines and Healthcare Products Regulatory Agency (MHRA), Bayesian confidence propagation neural network (BCPNN), and multi-item gamma Poisson shrinker (MGPS), and the criteria for positive results of each method are shown in Table 2.23–26 The satisfaction of all four conditions was considered a significant safety signal, which helped in reducing false-positive signals.
Two-by-two contingency table for disproportionality analysis.

AE, adverse event.
Formulas of four statistical analysis methods and their signal-generating-satisfied conditions.
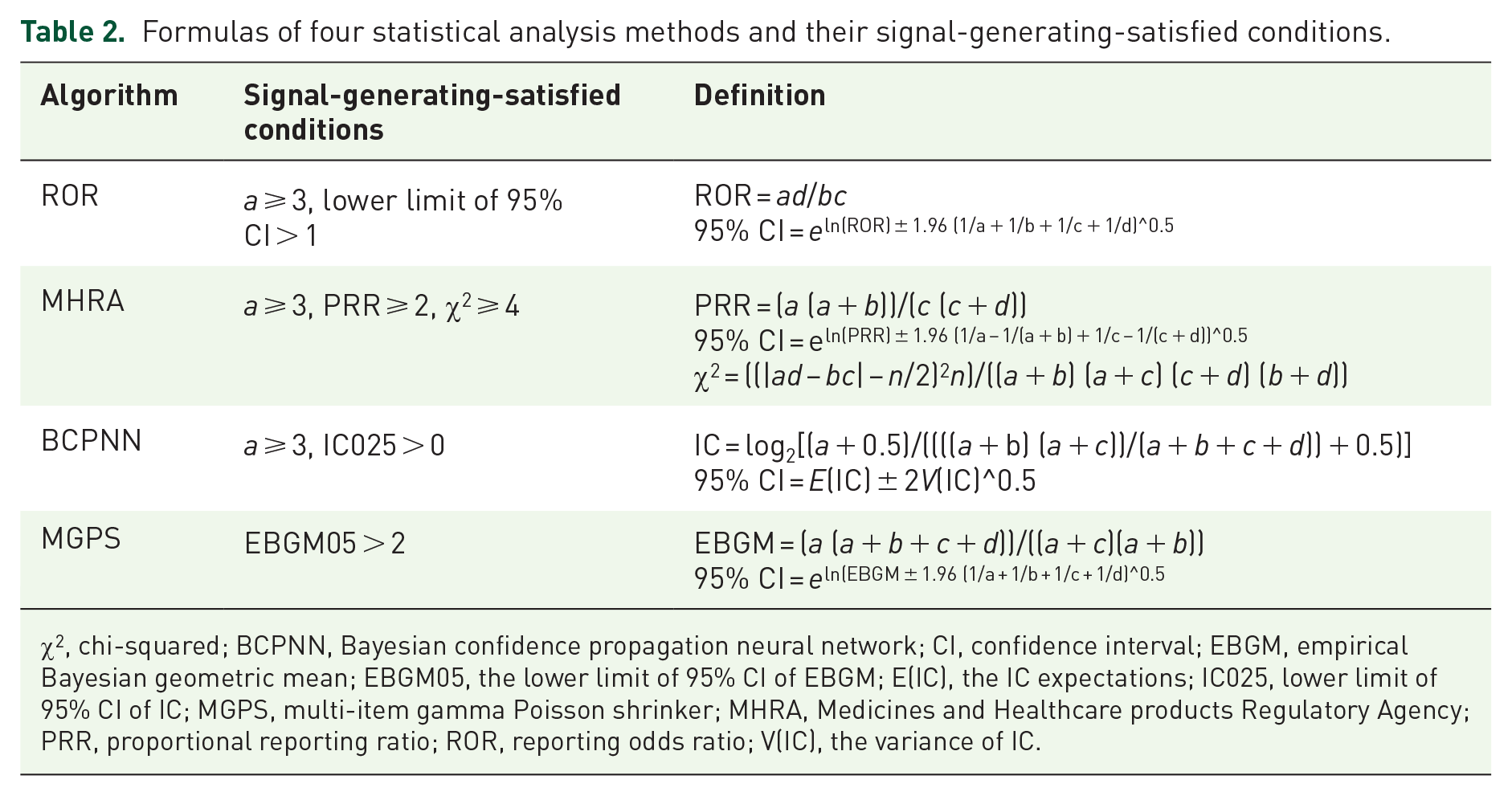
χ2, chi-squared; BCPNN, Bayesian confidence propagation neural network; CI, confidence interval; EBGM, empirical Bayesian geometric mean; EBGM05, the lower limit of 95% CI of EBGM; E(IC), the IC expectations; IC025, lower limit of 95% CI of IC; MGPS, multi-item gamma Poisson shrinker; MHRA, Medicines and Healthcare products Regulatory Agency; PRR, proportional reporting ratio; ROR, reporting odds ratio; V(IC), the variance of IC.
Outcomes of reports were classified as either serious or nonserious in FAERS at the time of reporting. SAEs were defined as those fatal, life-threatening, resulting in hospitalization, causing disability or permanent impairment, leading to congenital anomalies or birth defects, requiring intervention to prevent damage or permanent impairment, or other serious AEs. 27 Data has been cleaned by integrating the duplicate information, excluding unreported and unavailable data, and removing entries that were obviously inconsistent with established facts, to ensure its purity. TTO was calculated as the interval from the start of ribociclib to the date the AR occurred. Risk factors and PT signals of both serious and non-SAEs were examined by SPSS (version 22.0; IBM Corp., Armonk, NY, USA). In terms of exploring the relationship between drug dose and AEs, we excluded cases with unregistered dosage or dosing information clearly inconsistent with the reality (e.g., 125 mg/d), focusing only on patients who received 600, 400, or 200 mg/day ribociclib. Pearson χ2, Wilcoxon, Pearson χ2 Yates’s correction for continuity was applied according to data type, with statistical significance set at p < 0.05.
The reporting of this study conforms to the STROBE statement (Supplemental Table 2). 28
Results
Descriptive analysis
A total of 12,885 AE reports related to ribociclib were enrolled after deduplication. As shown in Table 3, 97.95% of the patients enrolled were female, with an average age of 60.30 ± 13.70 years. From 2017 to 2022, there was a steady increase in the number of AE reports associated with ribociclib. Regarding the outcome of AE, 25.73% of cases were classified as serious. With respect to TTO, 37.14% of patients experienced AEs within 30 days of starting ribociclib treatment, and 11.67% experienced them between 31 and 60 days. Besides, 18.52% of AEs were observed over 360 days.
Clinical characteristics of reports with ribociclib as the primary suspected drug from the FAERS database (January 2017 to June 2023).
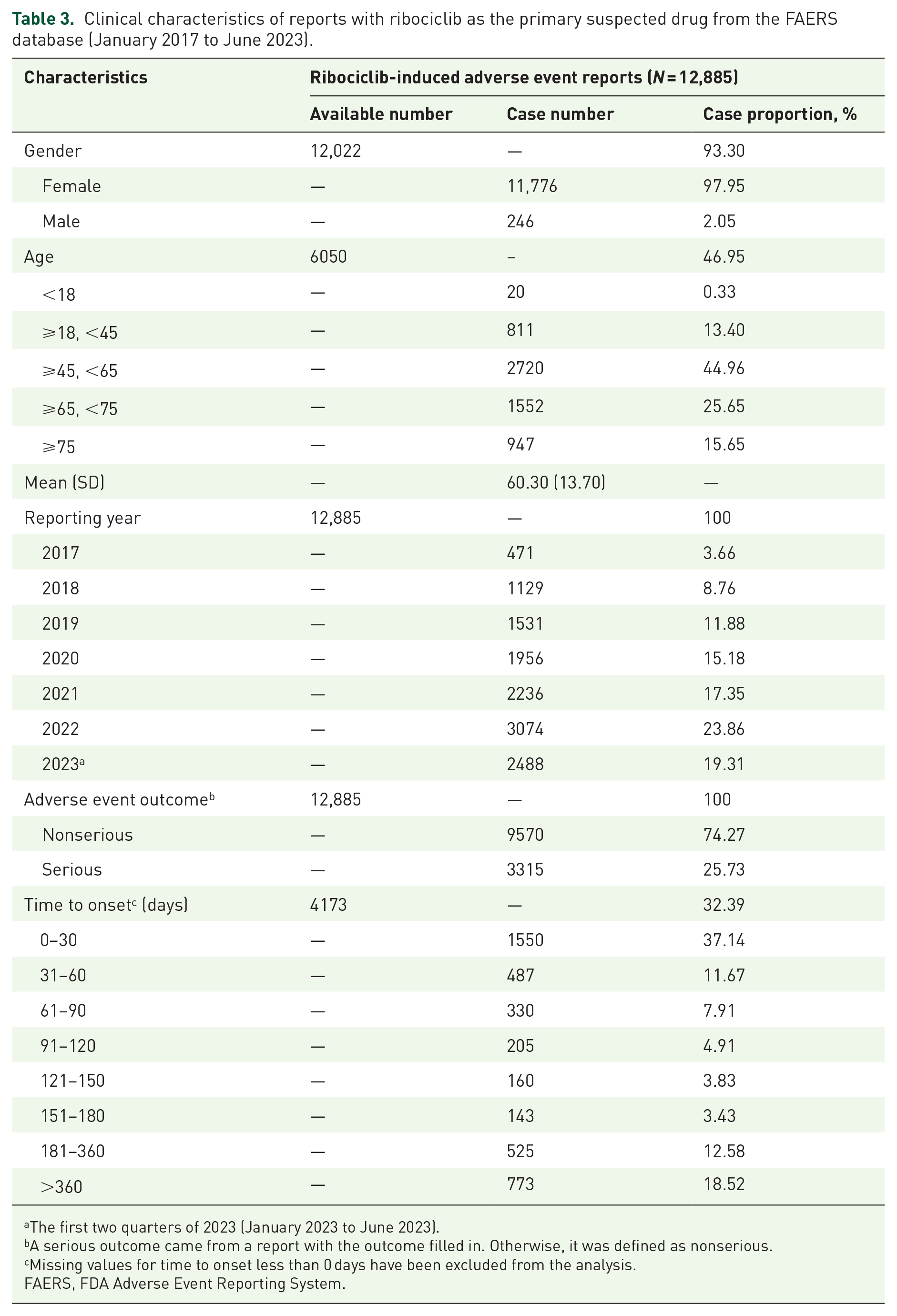
The first two quarters of 2023 (January 2023 to June 2023).
A serious outcome came from a report with the outcome filled in. Otherwise, it was defined as nonserious.
Missing values for time to onset less than 0 days have been excluded from the analysis.
FAERS, FDA Adverse Event Reporting System.
Disproportionality analysis
Signal at the SOC level
Signal strengths and corresponding reports at the SOCs level are detailed in Table 4. Among the SOCs, eight exhibited positive signals, achieving statistical significance in at least one of the four assessment methods. Three of them, respectively investigations (defined as AEs taking the form of abnormal laboratory tests; ROR: 2.24, 95% CI: 2.19–2.3; MHRA: 2.09, χ2: 4091.24; BCPNN: 1.06, IC025: 1.02; empirical Bayesian geometric mean (EBGM): 2.09, EBGM05: 2.03), neoplasms benign, malignant and unspecified (incl. cysts and polyps; ROR: 2.38, 95% CI: 2.31–2.45; MHRA: 2.27, χ2: 3219.98; BCPNN: 1.18, IC025: 1.13; EBGM: 2.27, EBGM05: 2.2), and blood and lymphatic system disorders (ROR: 2.7, 95% CI: 2.59–2.81; MHRA: 2.63, χ2: 2452.03; BCPNN: 1.39, IC025: 1.33; EBGM: 2.62, EBGM05: 2.51) showed statistically significant signals across all four methods used. Hepatobiliary disorders met three of them, whereas gastrointestinal disorders, skin and subcutaneous tissue disorders, respiratory, thoracic, and mediastinal disorders, and metabolism and nutrition disorders matched two. Thereinto, the signals observed for neoplasms benign, malignant, and unspecified (including cysts and polyps) might be influenced by the underlying progression of the disease itself rather than ribociclib.
Signal strength of reports of ribociclib at the SOC level in the FAERS database.

Indicates statistically significant signals in algorithm.
χ2, chi-squared; BCPNN, Bayesian confidence propagation neural network; CI, confidence interval; EBGM, empirical Bayesian geometric mean; EBGM05, the lower limit of 95% CI of EBGM; FAERS, FDA Adverse Event Reporting System; MHRA, Medicines and Healthcare products Regulatory Agency; PRR, proportional reporting ratio; ROR, reporting odds ratio; SOC, system organ class.
Signal at the PT level
A total of 68 PTs of ribociclib across 14 SOCs manifested statistically significant signals in all four methods (Supplemental Table 1). The most commonly reported AEs included nausea (n = 1426), neutropenia (n = 940), vomiting (n = 863), white blood cell count decreased (n = 812), and alopecia (n = 536), as listed in Table 5. Interestingly, our data mining revealed 28 unexpected AEs undiscovered in the drug label. Notable examples include skin discoloration (n = 106), hot flush (n = 155), and blood calcium decreased (n = 50).
Signal strength of reports of ribociclib at the PT level in the FAERS database.

Unexpected AEs.
χ2, chi-squared; AE, adverse event; BCPNN, Bayesian confidence propagation neural network; CI, confidence interval; EBGM, empirical Bayesian geometric mean; EBGM05, the lower limit of 95% CI of EBGM; FAERS, FDA Adverse Event Reporting System; MHRA, Medicines and Healthcare products Regulatory Agency; PT, preferred term; ROR, reporting odds ratio.
Serious versus nonserious cases
There were statistically significant differences between serious and nonserious AE outcomes in terms of age (60.07 years vs 61.83 years, p = 0.003) and body weight (69.72 kg vs 81.11 kg, p < 0.001), while no significant difference existed in gender distribution between the two groups, as shown in Table 6. Forty-three specific AEs that presented statistically significantly more serious reports than nonserious reports were identified (p < 0.05), such as neutropenia, anemia, gastritis, and pulmonary edema. (Table 7). It is particularly noteworthy that all cases of hepatotoxicity (n = 102) and asphyxia (n = 28) were classified as serious.
Differences in clinical characteristics between serious and nonserious reports with ribociclib as the primary suspected drug from the FAERS database.

Statistically significant (p < 0.05).
FAERS, FDA Adverse Event Reporting System.
Differences in PTs level between serious and nonserious reports with ribociclib as the primary suspected drug from the FAERS database.

Statistically significant (p < 0.05).
FAERS, FDA Adverse Event Reporting System; PT, preferred term.
The recommended starting dose for ribociclib is 600 mg/day, which strikes a balance between safety concerns and beneficial clinical action. 29 An initial reduction to 400 mg/day and a second reduction to 200 mg/day occur if dose modification is required. 30 As anticipated, most patients receiving the initial 600 mg/day dose experienced a notably greater number of AEs than those in other groups (Figure 2). When the dose was lowered from 600 to 400 mg/day, the data similarly demonstrated a significant drop in SAEs (Yates’s correction for continuity, χ2 = 67.368, p < 0.001). Nevertheless, there was no significant difference between patients receiving 400 mg/day and those getting 200 mg/day.

Doses of ribociclib used and number of cases reported or AEs (serious and nonserious).
Discussion
The official drug labeling is based on relatively tiny clinical trial populations, notwithstanding its authority. This post-marketing real-world study after drug marketing of AEs of CDK4/6 inhibitor focuses solely on ribociclib on ribociclib and offers more comprehensive and multifaceted information, even insights into rare AEs that may not have been captured before. We also analyzed clinical characteristics or PTs based on clinical outcomes, providing valuable guidance for physicians or pharmacists in clinical practice.
During the descriptive analysis, we noticed that 37.14% of AEs occurred within the first 30 days of starting ribociclib, and 11.67% occurred between 31 and 60 days. This pattern indicates that AE monitoring should be especially attentive during the initial stages of treatment. Additionally, 18.52% of AEs were recorded over a period of 360 days, which suggests the presence of long-latency AEs beyond our current understanding and highlights the potential for ribociclib to have long-lasting systemic adverse effects. This underscores the importance of sustained monitoring even long after the initiation of ribociclib therapy.
Nausea, reported as an AE, ranked first at the PT level. Nausea with serious outcomes ranked second among SAEs (1.87%), consistent with the result of the multicenter, phase IIIb RIBECCA study (1.8%). 31 However, it is irrational to conclude that nausea is a major AE for ribociclib with a poor prognosis. The high incidence of nausea among SAE is mainly due to the high incidence of nausea as an AE, which, in a statistically significant sense, mostly leads to nonserious outcomes. The well-known gastrointestinal toxicity of CDK4/6 inhibitors is more from abemaciclib rather than ribociclib, owing to the former’s activity at secondary pharmacologic targets instead of CDK4/6 inhibition. 32 In contrast, neutropenia, the second most common AE and the leading AE with serious outcomes was found to result in more serious outcomes than previously recognized. While the occurrence rate of SAEs due to neutropenia was reported to be less than 1% in the MONALEESA-7 trial, 33 our analysis of the FAERS database revealed a statistically significant rate of 1.92%, notably higher than in prior studies. This discrepancy raises a concern that the severity of neutropenia may have been underestimated.
As our study pointed out, skin and subcutaneous tissue disorders (e.g., alopecia, skin discoloration, and nail disorder) generally do not bring about serious outcomes. Within this category, special attention should be given to radiation dermatitis and radiation recall dermatitis. 34 Previous studies have revealed that when CDK4/6 inhibitors are administered simultaneously with radiotherapy, a higher percentage of cells may be in the G2/M phase of the cell cycle, which is more sensitive to radiation. As a result, it is recommended to avoid taking ribociclib when undergoing radiotherapy. 35 Besides radiation, light may also cause dermatitis linked to ribociclib. There have been documented cases of photosensitivity dermatitis as AE of ribociclib.36,37
Hepatobiliary disorders demonstrated significant signals at the SOC level in three methods, plus the rate of Grade 3/4 hepatobiliary toxicity exceeded 10% in all three MONALEESA trials, indicating hepatobiliary toxicity as an AE to reckon with. Our study found that apart from the transaminase elevations highlighted in the label, liver disorder, jaundice, and an unexpected AE hepatomegaly were also statistically significant. Although it is generally believed that ribociclib-reduced hepatobiliary toxicity is asymptomatic, does not lead to liver necrosis, and can be ameliorated by drug interruption, all reports of hepatotoxicity at the PT level in the FAERS database resulted in serious outcomes. Furthermore, cases of severe DILI and fulminant hepatitis that progressed even after discontinuation of ribociclib have been reported, reinforcing the credibility of our study’s findings.11,38 Patients with a history of alcoholism, hepatitis, and other hepatotoxic drugs deserve close monitoring. 39
A total of 28 unexpected and unlabeled AEs were spotted. Although strong positive signals were observed when ribociclib was identified as the primary suspected drug, blood calcium decreased, bone lesions, musculoskeletal chest pain, bone disorder, and polyneuropathy are generally recognized as common AEs associated with concomitant drugs, especially aromatase inhibitors and chemotherapeutic agents. In addition, pleural effusion and ascites may be attributable to the underlying tumor pathology itself. ILD, which carries an FDA black box warning, is well-recognized as an SAE with a lengthy latency (about 253 days under the recommended daily dose 40 ). However, according to our research, there are other unidentified AEs that fall under the category of respiratory, thoracic, and mediastinal disorders and have the potential to be extremely harmful. These include pulmonary pain, pulmonary edema, and pulmonary mass. Two deaths of dyspnea and pneumonia considered related to ribociclib were reported. 41 Although the mechanism by which ribociclib contributes to these unlabeled AEs remains largely unexplored, it is encouraged to add them to the drug label so that patients and healthcare providers can be aware of these potential risks in clinical practice, detect and intervene them in time to avoid the occurrence of SAEs.
Dose reduction of ribociclib, when medically necessary, has been reported to alleviate certain AEs emerging during treatment.42,43 The overall incidence of AEs is numerically lower in real-world clinical practice compared to clinical trials, while the frequency of dose reduction due to AEs is higher. An observational retrospective study revealed that reducing the ribociclib dose did not adversely impact its efficacy. 44 This suggests a positive attitude to dose reduction in response to AEs. While dose reduction is a valuable strategy for managing AEs, however, we found that further dose reduction below 400 mg/day may not substantially decrease the frequency of AEs or SAEs. However, due to the existence of a high rate of missing data for dosages of ribociclib in the FAERS database, clinicians should flexibly assess the need for dose adjustment based on the general condition and AEs of different patients in clinical practice.
Limitations
Our study has several unavoidable limitations. First, the FAERS database, as an open system, naturally suffers from reporting and data biases. More than a third of the reports registered occurred in the United States, and not all countries have approved ribociclib for early stage breast cancer. Second, disproportionality analysis does not establish direct causal relationships. The absence of control groups or exclusion of potential confounding factors in our analysis means that follow-up research based on long-term data or further prospective studies is necessary to validate the association and reveal the causality between a specific AE, especially unlabeled AE, and ribociclib. Third, observational designs such as this study cannot fully rule out all sources of confounding. The original diagnosis when an AE occurs is not available in the FAERS database. Consequently, the potential differences in the AEs associated with various indications remain to be elucidated. Due to the large sample size and the limitations posed by incomplete baseline information of patients in the FAERS database, despite statistical significance, whether age and body weight have an independent impact on AE outcomes of ribociclib still requires further investigation realized by studies with more comprehensive and detailed datasets.
Conclusion
Our study aimed to identify real-world safety signals of ribociclib by analyzing the AEs after its marketing. We found that AEs mainly occurred in the first 60 days of ribociclib administration. Blood and lymphatic system disorders and abnormalities in investigation seemed to be strongly related to ribociclib. Nausea, neutropenia, vomiting, white blood cell count decreased, and alopecia turned out to be the most commonly reported AEs. Twenty-eight unlabeled AEs were newly identified, raising concern for wider clinical monitoring and further investigation of the causality. As a widely recognized AE, neutropenia was revealed to possibly lead to more serious outcomes than expected. Follow-up research and long-term data are necessary to confirm our results detected from the FAERS database, and our study can serve as a warning to clinicians and provide a direction for further studies.
Supplemental Material
sj-docx-1-taw-10.1177_20420986251324633 – Supplemental material for A post-marketing disproportionality analysis of the safety of ribociclib based on the FDA Adverse Event Reporting System
Supplemental material, sj-docx-1-taw-10.1177_20420986251324633 for A post-marketing disproportionality analysis of the safety of ribociclib based on the FDA Adverse Event Reporting System by Jiayan Xu, Ruo Wang and Kunwei Shen in Therapeutic Advances in Drug Safety
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.