
Abstract
Introduction:
The drug pharmacovigilance system in Japan is similar to those in the European Union (EU) and the United States. As a unique Japanese pharmacovigilance program, postmarketing all-case surveillance (PMACS) is required. PMACS plays a key role for postmarketing activities, but there are challenges that place much burden on PMACS conduct. This study investigates the impact of PMACS on postmarketing activities in Japan and proposes its potential improvement. This study also seeks the possibility to expand PMACS beyond Japan.
Materials and Methods:
Reexamination reports issued from 2017 to 2019 were identified in September 2020 by searching ‘reexamination report’ and ‘201701’ to ‘201912’ on the Pharmaceuticals and Medical Devices Agency website. The corresponding Package Insert (PI) change orders and premarketing review reports were also identified. Reviewing these regulatory documents allowed for investigation of the PMACS impact on postmarketing activities.
Results:
More than half (57%) of the drugs with PMACS had ‘Limited dosing experience in Japan’ as a reason for the PMACS requirement. As a safety measure, no PI change orders were imposed on 33% and 28% of drugs with and without PMACS, respectively. The means of the number of PI change orders were 2.23 and 2.14 for drugs with and without PMACS, respectively. There were no reexamination reports mentioning any concerns related to efficacy.
Discussion and Conclusion:
PMACS should not be imposed only because of limited dosing experience in Japan at the premarketing stage. Rather, PMACS should focus on (1) collection of safety data (not efficacy), (2) necessity of distribution control, and/or (3) collection of case details for drugs with a limited treated population. PMACS also has the potential to be utilized in the EU and the United States, as their regulatory frameworks are acceptable for PMACS. Naglazyme (galsulfase) is a case where the PMACS-like studies have been required in each region.
Plain Language Summary
Effectiveness of data collection for all patients who receive a new drug as a safety measure in Japan
Introduction:
In Japan, a drug company is obligated to conduct data collection after a new drug launch as an approval condition. The obligation is a unique Japanese requirement where a company must collect data from all patients receiving the drug in Japan in cooperation with hospitals. This is expected to contribute to intensive data collection and better drug distribution control and could potentially be useful in countries beyond Japan. However, no clear criteria have been established for decision making, despite the significant burden for companies and hospitals. Therefore, this study aimed to investigate the impact of the obligation on safety measures and efficacy data collection and propose a potentially improved drug scope to impose the obligation.
Materials and Methods:
Reexamination of reports issued by the Pharmaceuticals and Medical Devices Agency between 2017–2019.
Results:
More than half (57%) of the included drugs had ‘Limited dosing experience in Japan’ as a reason for the obligation being required. However, regulatory order to change drug label, an action based on safety signal identification, was imposed on 33% and 28% of drugs with and without the obligation, respectively. The means of the number of the label change orders were 2.23 and 2.14 for drugs with and without obligation, respectively. Meanwhile, some drugs were highlighted as potential factors for better application of the obligation.
Conclusion:
According to these results, the obligation should be imposed on a limited number of drugs by focusing not on dosing experience in Japan but on safety (not efficacy) data collection, necessity of distribution control, and/or collection of case details for drugs with a limited treated population. The obligation also has the potential to be utilized in the EU and the United States, as their regulatory frameworks are acceptable for the obligation.
Introduction
The drug pharmacovigilance system in Japan has three key components: (A) adverse reaction reporting system, (B) reexamination system with a Periodic Safety Update Report (PSUR), and (C) postmarketing study/surveillance. 1 Figure 1 shows an overview of the pharmacovigilance system in Japan.

Pharmacovigilance system for Drugs in Japan. Adverse reaction reporting system, reexamination system and postmarketing study/surveillance are a key driver for drug pharmacovigilance system in Japan.
The pharmacovigilance system in Japan is similar to those in the European Union (EU) and the United States:
A. Adverse reaction reporting system: The Japanese regulators, the Ministry of Health, Labour and Welfare (MHLW) and the Pharmaceuticals and Medical Devices Agency (PMDA), monitor and assess safety signals, and take regulatory measures as needed. Similar systems for adverse reaction reporting have been implemented in the EU (pharmacovigilance system) 2 and the United States (FDA adverse event reporting system). 3
B. Reexamination system with PSUR: The reexamination system in Japan requires a Marketing Authorization Holder (MAH) to collect data for drugs and prepare PSURs for a certain period in their postmarketing stage. At the end of the reexamination period, the Japanese regulators assess the benefit/risk balance of these drugs. Systems to collect postmarketing data, prepare PSURs and assess the benefit/risk balance of an approved drug have been implemented in the EU (PSURs) 4 and the United States (Periodic adverse drug experience reports) 5 as well.
C. Postmarketing study/surveillance: A postmarketing study/surveillance requirement/commitment is imposed on an MAH upon drug approval in Japan. The EU and the United States have similar regulatory systems, Post-Authorisation Safety Studies (PASS) 6 and Postmarketing Requirements and Commitments, 7 respectively.
However, as a unique Japanese pharmacovigilance requirement, the MHLW is authorized to compel an MAH to conduct a postmarketing all-case surveillance (PMACS) as a drug approval condition. PMACS is a type of postmarketing study/surveillance requirement/commitment mentioned above. PMACS is an observational single-arm postmarketing study where an MAH must work with medical institutes to collect data from all the patients received its drug in Japan. PMACS has been implemented for more than 20 years in Japan, 8 and the number of approved drugs with PMACS as an approval condition have increased since 2003. 9
The MHLW explains in the PMACS notification that PMACS is obligatory in the cases where the number of subjects in Japan is low or none in clinical studies for a drug at the premarketing phase or where the drug is likely to cause serious adverse effects. 10 The MHLW expects that PMACS contributes to collecting demographic information on patients in Japan and information on safety and efficacy quickly and without any biases.
MAHs with PMACS experience indicated that PMACS had been useful mainly because (a) case details had been collected even in a limited population, (b) information on off-label use had been obtained, and (c) eligibility of medical institutes, physicians, and patients had been confirmed. 11 Meanwhile, it has been reported that the resource burden for MAH to conduct PMACS was more than 1.5 times (more than 5.0 times in some surveillances) compared with general postmarketing activities, including postmarketing studies. 11 Moreover, PMACS was burdensome for medical institutes due to excessive data collection and entity in a specified form, complicated survey form use, cumbersome contract procedures, and long-term patient follow-up.
Therefore, whether to impose PMACS or not as an approval condition should be carefully decided, taken into account the balance between expected achievement and necessary burden. However, there exist no specific criteria for the decision. Approximately 90% of drugs with PMACS have the reason for its obligation that the number of Japanese patients in the premarketing phase was too small. 11
In summary, PMACS has played a key role in the pharmacovigilance system for drugs in Japan, while the decision whether PMACS is required or not was mainly made by limited dosing experience in Japan, in spite of the high burden of its conduct on MAH and medical institute. This study aimed to investigate the impact of PMACS on safety measures and efficacy data collection in Japan. According to the investigation, we propose a possible improvement of the product scope to impose PMACS as a drug approval condition. The proposal contributes to better pharmacovigilance activities in Japan. This study also considers the potential to introduce PMACS beyond Japan based on the pharmacovigilance systems in Japan, the EU, and the United States.
Materials and methods
Data material sources
There are three types of Japanese regulatory documents used in this study; (A) drug reexamination reports, (B) drug premarketing review reports, and (C) Package Insert (PI) change orders.
A. Drug reexamination report: It is prepared and published by the PMDA once the reexamination period has ended. The report summarizes the information related to safety and efficacy data collected during the specified reexamination period. This includes PMACS data, if applicable.
B. Drug premarketing review report: It is prepared and published by the PMDA once the drug is approved. The report includes information on the approval conditions, including PMACS requirements.
C. PI change order: It is a regulatory notification issued by the MHLW as a safety measure at the postmarketing stage of a drug. PI is equivalent to Summary of Product Characteristics (SmPC) in EU and drug label in the United States. The MHLW issues the order if the PMDA identifies a safety signal for the drug and completes the assessment based on accumulated safety data via the adverse reaction reporting system, the PSUR, and postmarketing study/surveillance, including PMACS. An MAH must change the drug PI, according to the notification.
These documents can be found in the PMDA website to search for information on each drug in Japan. 12 In this study, the reexamination reports issued from 2017 to 2019 were identified in September 2020 by searching for ‘reexamination report’ and ‘201701’ to ‘201912’ on the PMDA website. The corresponding PI change orders and premarketing review reports were also identified on the same website.
The 3-year period is expected to present the trend of deciding whether to impose PMACS after the PMDA started conducting drug premarket reviews. The PMDA was established in April 2004, 13 and the standard duration of the premarketing priority review by the PMDA has been set at 9.0 months. 14 Therefore, new drugs reviewed by the PMDA were launched from 2005. The reexamination period of a drug is up to 10 years. 15 The median assessment duration for reexamination by the PMDA was 15.0 months in Fiscal Year 2018. 14 Hence, the reexamination reports from 2017 are expected to cover the majority of drugs reviewed by the PMDA.
Method to investigate the impact of PMACS
Listing and categorization of reexamination report
Reexamination reports issued from 2017 to 2019 were listed with issue date, nonproprietary name and brand name. Each report was flagged with the items below by checking premarketing review reports of these drugs:
A. Approval type: Identify whether the approval is for a new drug or label expansion (e.g. new indication).
B. PMACS: Identify whether PMACS was conducted.
C. Reason for PMACS: Categorized the reasons for PMACS with ‘Limited dosing experience in Japan’, ‘Further data collection’, ‘Specific safety concern(s)’, or ‘Other’.
Then, the items below were added to the list by investigating PI change orders of each drug:
D. PI change order: Identify whether the MHLW issued order(s) for PI change during the reexamination period.
E. Number of PI change orders: Enter the number of PI change orders for each drug during the reexamination period.
Based on the list with these items above, the reexamination reports were categorized by (A) Approval type, (B) PMACS, and (D) PI change order. In addition, the number of reexamination reports were compared by (C) Reason for PMACS.
Assessment from viewpoint of drug safety measures
The number of PI change orders for each drug were checked. The number is considered a quantitative index for safety measures taken during their reexamination period, based on the description of PI change order in Part C of the section ‘Data material sources’.
Then, drugs with five or more PI change orders issued during their reexamination periods were categorized by therapeutic area of the World Health Organization Anatomical Therapeutic Chemical and Defined Daily Dose (WHO ATC/DDD) index. Five or more PI change orders means that safety measures were taken every 2 years or more frequently on average during reexamination period.
Assessment from viewpoint of drug efficacy data collection
The efficacy information in each reexamination report was examined to identify any specific findings related to efficacy.
Identification of distinctive cases in term of PMACS impact
Each reexamination report was reviewed to identify any distinctive drugs/indications where decision with PMACS/no PMACS affected their postmarketing activities.
Results of investigation for the impact of PMACS
Listing and categorization of reexamination report
According to the search using the conditions outlined in the section ‘Data material sources’, 221 reexamination reports were identified in 2017–2019. Two reports for the egg-derived influenza A (H5 N1) vaccine were excluded from this study, because these vaccines had not been marketed during the reexamination period. In addition, a report on sitagliptin was excluded as two similar reports with two different brand names were published due to concurrent selling by two MAHs in Japan. Therefore, 218 reports were included in this study in total. The list of these reports is shown in Table 1.
List of reexamination reports issued from 2017 to 2019.

NA, not applicable; PI, package insert; PMACS, postmarketing all-case surveillance.
The numbers in this column mean 1: Limited dosing experience in Japan; 2: Further data collection; 3: Specific safety concerns; 4: Other.
Figure 2 shows the categorization of the 218 reexamination reports. Among them, 158 and 60 cases were for new drug and label expansion, respectively. PMACS was obligatory in 52 of the 158 cases with new drug approval. Among the 52 cases, one or more PI change orders were issued in 36 cases (69%) during the reexamination period. Meanwhile, there were 106 cases with new drug approvals where PMACS was not obligatory. Among the 106 cases, one or more PI change orders were issued in 73 cases (69%) during their reexamination periods.
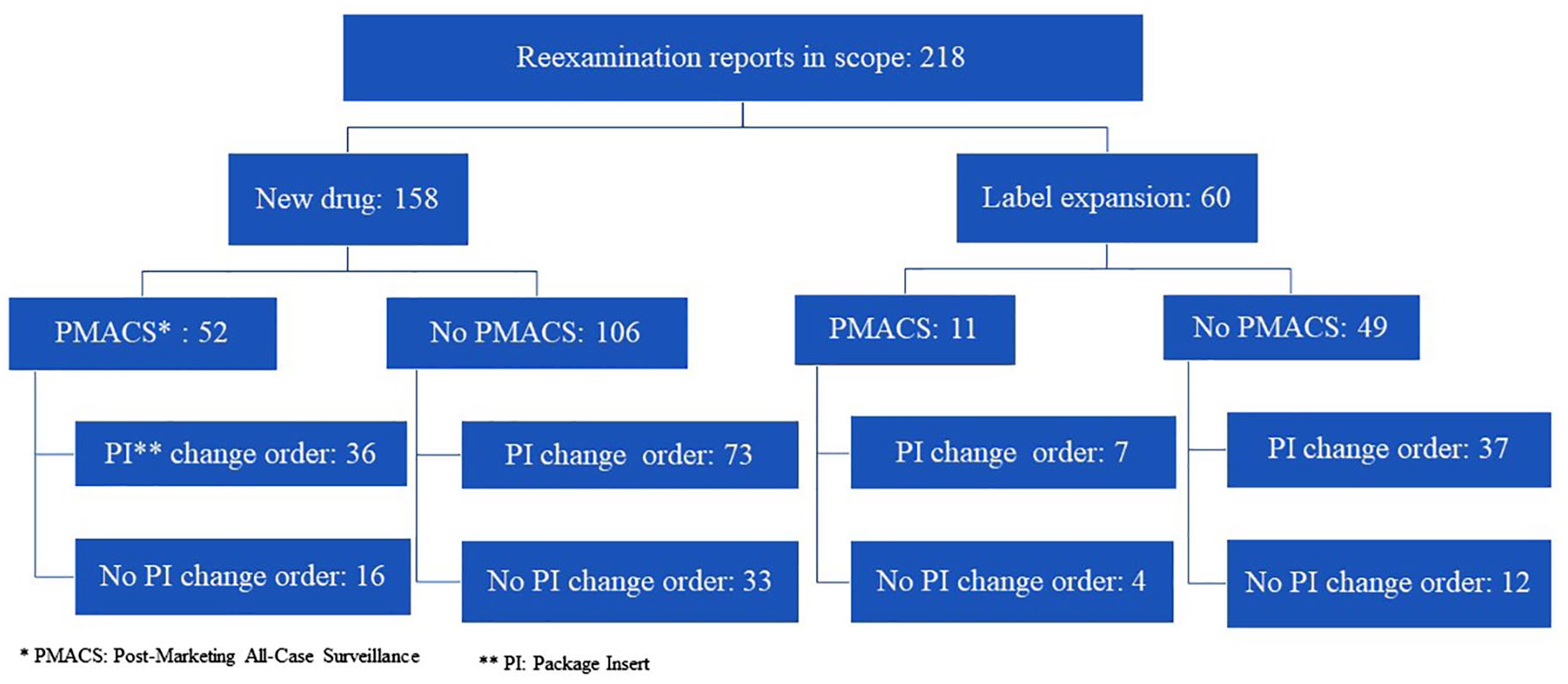
Reexamination report categorization. The identified 218 reexamination reports in 2017–2019 were categorized by PMACS requirement and PI change order, and the breakdown number of reexamination reports in each category is shown.
PMACS was obligatory in 11 of the 60 cases with label expansion. One or more PI change orders were issued in 7 (64%) of the 11 cases during their reexamination periods. Meanwhile, there were 49 cases with label expansion where PMACS was not obligatory. Among the 49 cases, one or more PI change orders were issued in 37 cases (76%) during the reexamination period.
Figure 3 compares the number of reexamination reports by reason for PMACS. More than half (57%) of the cases with PMACS had ‘Limited dosing experience in Japan’ as a reason for PMACS obligation. PMACS was conducted voluntarily in four out of five cases with the reason ‘Other’. The fifth case was Tracleer (bosentan hydrate) (Janssen Pharmaceutical K.K., Japan), for which PMACS was required because the indications approved were wider than those targeted in premarketing clinical studies.
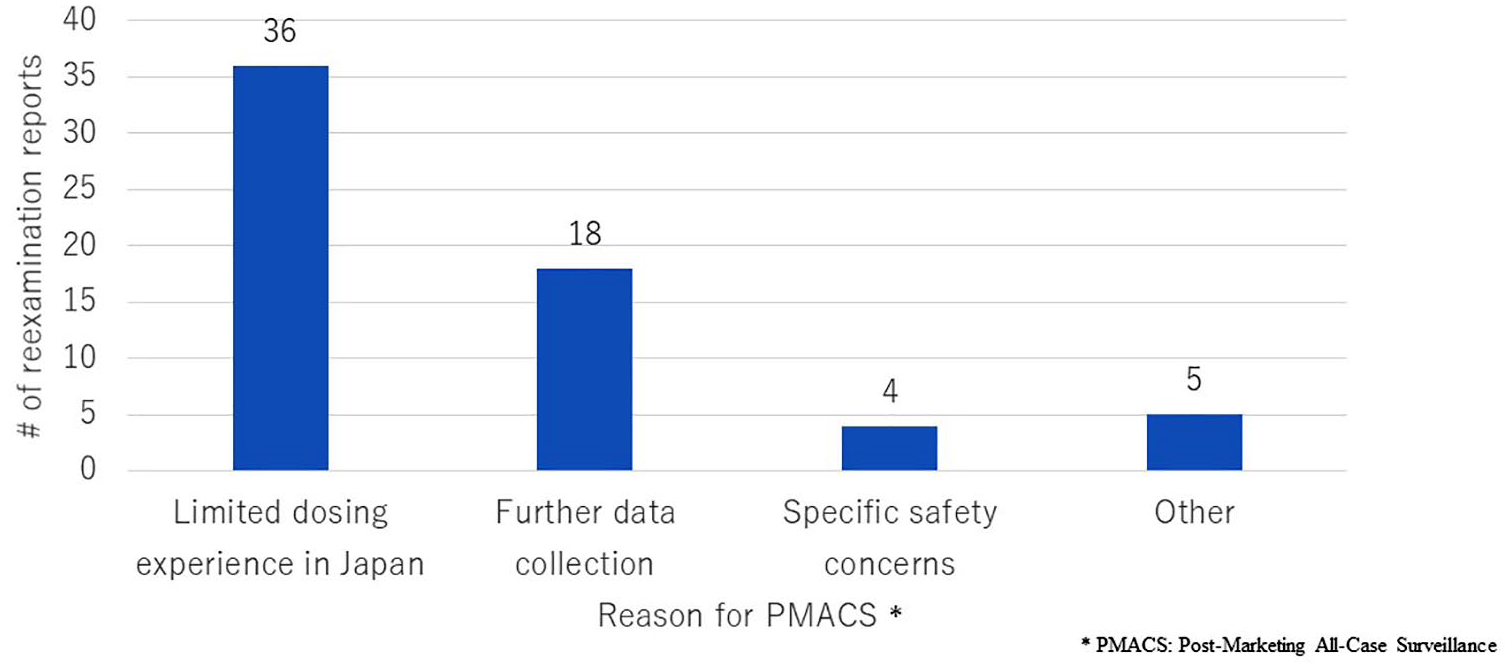
Number of reexamination reports by reason for PMACS. The number of reexamination reports issued from 2017 to 2019 was compared by reason for PMACS.
Assessment from viewpoint of drug safety measures
The number of PI change orders was identified for 198 drugs, as two reexamination reports were issued for 20 drugs, since these reports targeted different indications. As a PI change order is issued for the relevant drug, not for each indication, these 20 reports were excluded from the calculated number to avoid duplicate counting. Table 2 summarizes these results. No PI change orders were issued for 33% and 29% of drugs with and without PMACS, respectively, during their reexamination periods. The means of the number of PI change orders were 2.23 and 2.15 for drugs with PMACS and without PMACS, respectively.
Number of PI change orders.
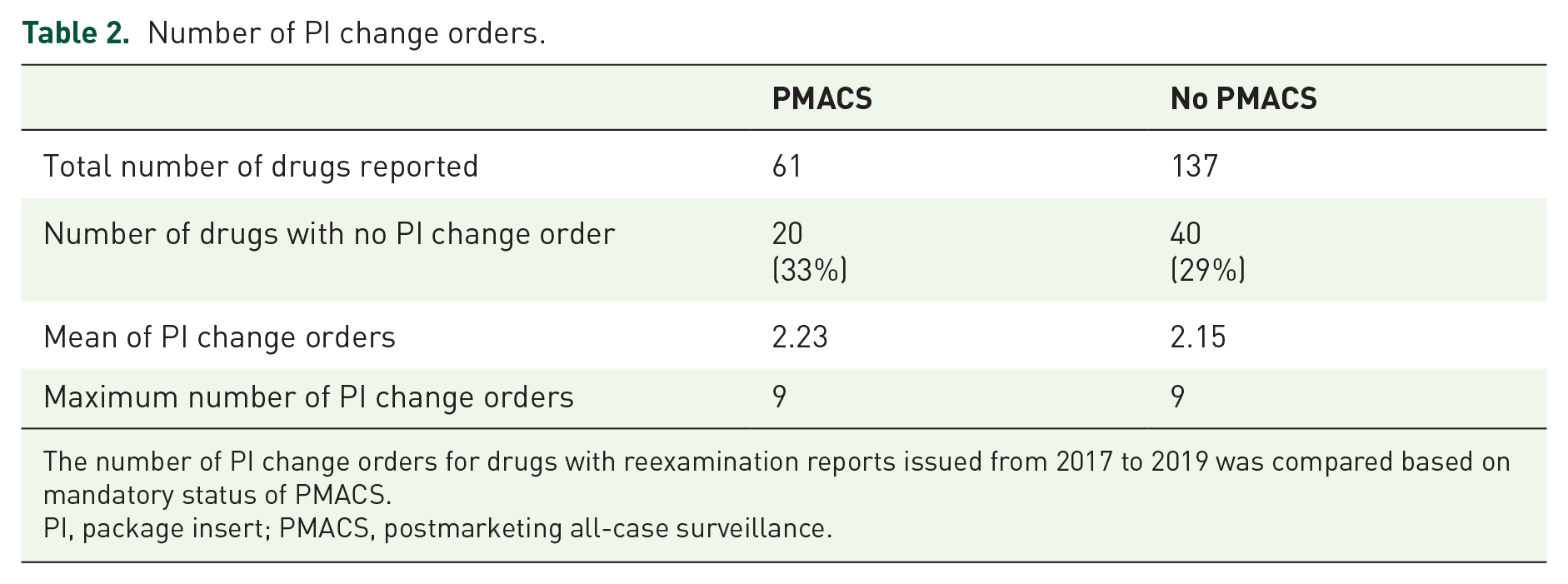
The number of PI change orders for drugs with reexamination reports issued from 2017 to 2019 was compared based on mandatory status of PMACS.
PI, package insert; PMACS, postmarketing all-case surveillance.
Table 3 shows the number of drugs with five or more PI change orders issued during their reexamination periods by WHO ATC/DDD index. Totally, five or more PI change orders were issued in 10 out of 61 (16%) drugs with PMACS and 20 out of 137 (15%) drugs without PMACS. The highest number of the drugs fell into ‘anti-infectives for systemic use’ (11 drugs). However, the number was obtained due to the PI change order, not for a specific product, but for all the influenza related products in Japan with four influenza vaccines and Tamiflu (oseltamivir phosphate) (Chugai Pharmaceutical Co., Ltd., Japan). Once the order is excluded, ‘anti-neoplastic and immunomodulating agents’ have the highest number (eight drugs).
Number of drugs with five or more PI change orders by WHO ATC/DDD index.

Among drugs with reexamination reports issued from 2017 to 2019, those with five or more PI change orders during their reexamination period were accumulated, categorized by WHO ATC/DDD index and compared based on mandatory status of PMAC.
PMACS, postmarketing all-case surveillance; WHO ATC/DDD, World Health Organization anatomical therapeutic chemical and defined daily dose.
Assessment from viewpoint of drug efficacy data collection
There were no reexamination reports mentioning any outstanding findings related to drug efficacy.
Identification of distinctive cases in term of PMACS impact
Four drugs were identified by review of each reexamination report in terms of decision with and without PMACS which affected postmarketing activities; Nexavar (sorafenib tosilate) (Bayer Yakuhin, Ltd, Japan), Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan), Lamictal (lamotrigine) (GlaxoSmithKline K.K., Japan) and Concerta (methylphenidate hydrochloride) (Janssen Pharmaceutical K. K., Japan).
Discussion
Listing and categorization of reexamination report
As Figure 3 shows that PMACS was obligatory in more than half (57%) of the cases due to ‘Limited dosing experience in Japan’, the MHLW has made a decision of PMACS/no PMACS, taking much into account dosing experience in Japan. However, ‘Limited dosing experience in Japan’ is not a suitable decision criterion, as no difference of safety measures or efficacy data collection was observed between the cases with PMACS and no PMACS in this study.
Another finding is that dosing experience with different indications in Japan is considered when deciding on the PMACS obligation. PMACS was obligatory for 52/158 cases in new drugs versus 11/60 cases in label expansions.
Assessment from viewpoint of drug safety measures
As shown in Table 2, similar percentages of drugs with no PI change were observed (33% and 29% of drugs with and without PMACS, respectively). In addition, the mean number of PI change orders was similar between drugs with PMACS and no PMACS. These results indicate that postmarketing activities achieved safety measures to a similar degree between with and without PMACS. Therefore, the decision of PMACS/no PMACS based on mainly ‘Limited dosing experience in Japan’ contributed little to safety measures while the PMACS burden occurred.
Assessment from viewpoint of drug efficacy data collection
As mentioned in the section ‘Assessment from viewpoint of drug efficacy data collection’, no outstanding efficacy findings were observed in reexamination process, regardless of with and without PMACS. This means that decision of PMACS/no PMACS have little impact on efficacy data collection, even though the PMACS was required mainly for the puropse of ‘Limited dosing experience in Japan’ or ‘Further data collection’. This point is also supported, considering that, as mentioned in the section ‘Introduction’, PMACS is usually a single-arm observational study where findings for efficacy are likely limited. In fact, postauthorization efficacy studies are separated from PASSs and clearly imposed, if needed, in the EU. 16
Identification of distinctive cases in term of PMACS impact
No distinctive cases were identified from the viewpoint of drug efficacy data collection, as there were no findings in any reexamination reports as mentioned in the section ‘Assessment from viewpoint of drug efficacy data collection’. The four drugs shown in the section ‘Identification of distinctive cases in term of PMACS impact’ were categorized as follows in terms of PMACS decision and safety measures taken:
A. Drug with PMACS, resulting in outstanding safety measures: Nexavar (sorafenib tosilate) (Bayer Yakuhin, Ltd, Japan) and Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan)
B. Drug without PMACS, resulting in outstanding safety measures: Lamictal (lamotrigine) (GlaxoSmithKline K.K., Japan)
C. Drug with PMACS, resulting in failed safety measures: None
D. Drug without PMACS, resulting in failed safety measures: Concerta (methylphenidate hydrochloride) (Janssen Pharmaceutical K. K., Japan)
Details of each case are explained below:
A. Nexavar (sorafenib tosilate) (Bayer Yakuhin, Ltd, Japan), an anticancer drug, could be considered one of the best examples where PMACS worked well. When it was approved, data from 171 Japanese patients had been accumulated in premarketing clinical studies. 15 The number of Japanese patients treated at premarketing stage was higher than the number of Japanese patients influencing the obligatory decision with PMACS (100 patients). 9 That is, the MAH would not have been obligated to conduct PMACS in term of limited dosing experience in Japan. However, PMACS was obligatory. This was because the drug was the first molecular-targeted drug in the field of urological malignancies in Japan and because a variety of adverse drug reactions were observed during premarketing clinical studies. 15 As a result, eight PI change orders were issued during the reexamination period, including two urgent safety information letters. 17 One of the letters was related to interstitial pneumonia, an unexpected adverse drug reaction that had not been observed in the premarketing phase. The other was related to liver failure and hepatic encephalopathy. Liver failure was one of the safety notes highlighted by PMACS, according to data from premarketing clinical studies. PMACS contributed to the detection of both known and unknown safety concerns for this drug, regardless of dosing experience in Japanese patients.
Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan) is also considered appropriate to apply PMACS. Only seven patients were treated with this drug for more than a 10-year PMACS period. 18 No new important safety concerns were observed, and efficacy information was limited because of the low number of patients. However, data from all patients were intensively collected for a long period under PMACS. The data of each case could help the understanding of its use.
B. Lamictal (lamotrigine) (GlaxoSmithKline K.K., Japan) supports the idea that postmarketing activities without PMACS are effective. For this drug, the urgent safety signal regarding serious skin disorders and the underlying cause were identified without PMACS. Accordingly, some safety actions, including issuing an urgent safety information letter, 17 were taken to ensure dose compliance at the escalation phase and quick discontinuation of drug dosing if a skin disorder was observed. As a result, the number of cases of inappropriate use decreased.
C. There was no case identified in this category. In general, PMACS is a good tool to collect safety data comprehensively, as all patients treated with the targeted drug are captured.
D. Concerta (methylphenidate hydrochloride) (Janssen Pharmaceutical K.K., Japan) is considered a case where PMACS should have been required since its launch. The drug was not required to conduct PMACS as an approval condition, while supply restrictions, including prescribing physician registration, needed to be introduced. As a result, more than 500 prescriptions were issued by ineligible physicians during its reexamination period; the drug was dispensed based on 75 inappropriate prescriptions; and no serious adverse drug reactions due to the inappropriate prescriptions were observed. 19 Then, according to the reexamination assessment by the PMDA, further measures were required for its appropriate use, including the introduction of a system to register all patients to be prescribed.
Proposal on PMACS application
As discussed in sections ‘Listing and categorization of reexamination report,’ ‘Assessment from viewpoint of drug safety measures’, and ‘Assessment from viewpoint of drug efficacy data collection’, the decision of PMACS/no PMACS based on mainly ‘Limited dosing experience in Japan’ contributed little to safety measures or efficacy data collection, in spite of high PMACS burden. Meanwhile, the four distinctive cases are identified in terms of PMACS decision and safety measures taken. Based on these points, the proposed product scope to impose PMACS is as follows:
A. Drugs related to Designated Diseases in Article 67 of the Act on Securing Quality, Efficacy and Safety of Products Including Pharmaceuticals and Medical Devices (PMDA Act) 20 : currently, cancer, sarcoma, and leukemia are specified as a designated disease according to the article referring to drugs ‘for which use not under the guidance of physicians or dentists is highly likely to cause hazards.’
Nexavar (sorafenib tosilate) (Bayer Yakuhin, Ltd, Japan) supports this proposal. This case represents a successful application of mandatory PMACS, even though dosing experience in Japanese patients at the premarketing stage was available to some extent. In addition, as highlighted in Table 3, ‘antineoplastic and immunomodulating agents’ had the highest number (eight) of drugs with five or more PI change orders issued during their reexamination periods. This indicates that intensive safety information collection under PMACS contributed to advancing the appropriate use of drugs in the therapeutic area.
B. Drugs with special distribution restrictions: Concerta (methylphenidate hydrochloride) (Janssen Pharmaceutical K.K., Japan) supports this proposal. For example, the distribution of narcotics must be strictly managed in Japan to avoid drug abuse or dependence. For such a drug, PMACS would allow tracking appropriate drug use and collecting safety and efficacy data efficiently, because distribution restriction and data collection could be pursued simultaneously.
C. ‘Ultra-orphan’ drugs: There is no legal definition of ‘Ultra-orphan’ drugs in Japan and other countries/regions such as in the EU and the United States, but the Scottish Medicine Consortium has set a definition that considers the prevalence (1 in 50,000 or less) and conditions, including chronicity, severe disablement, and necessitating highly specialized management. 21 Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan) supports this proposal. For such a drug, PMACS could contribute to closely monitoring every patient and quickly collecting and sharing detailed information on each dosing experience for appropriate drug. Furthermore, the burden for MAHs and medical institutes to conduct PMACS could be reasonable because of the low number of patients to be followed up.
Finally, PMACS could be utilized not only in Japan but also in other regions/countries such as the EU and the United States. As described in the section ‘Introduction’, both regulators have a similar pharmacovigilance system similar to that in Japan with the authority to impose MAHs to conduct a postmarketing study. Thus, these systems already have the regulatory framework to introduce PMACS, enabling proposition by regulators and MAHs alike, if preferable.
For example, Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan) has been approved not only in Japan but also in the EU and the United States, and clinical surveillance programs have been conducted as a condition of its authorization/approval in the EU and the United States.22,23 One of the objectives of the programs was to collect clinical data from as many treated patients as possible. In this case, the programs could be appropriately conducted as PMACS, similar to that in Japan.
In addition, Nexavar (sorafenib tosilate) (Bayer Yakuhin, Ltd, Japan), which had PMACS obligation and resulted in outstanding safety measures, indicated that PMACS can be effective not only in Japan but also other countries. The reasons to decide PMACS obligation were not Japan-specific ones (e.g. ‘Limited dosing experience in Japan’).
Conclusion
This study revealed that the appropriate safety action (issuing PI change order) was taken, regardless of the mandatory status of PMACS and the degree of dosing experience in Japanese patients at the premarketing stage. Meanwhile, it also identified some drugs where PMACS worked well or could be effective. Considering the significant burden PMACS exerts on MAHs and medical institutes, in addition to these study findings, the product scope to impose PMACS should be improved. Particularly, PMACS should not be imposed only because of limited dosing experience in Japan at the premarketing stage. Rather, PMACS requirement should focus on (1) collection of safety data (not efficacy), (2) necessity of distribution control, and/or (3) collection of case details for drugs with a limited treated population. Finally, PMACS has the potential to be utilized not only in Japan but also in other countries/regions, such as the EU and the United States. Both regulators have similar pharmacovigilance systems to Japan, which means that they already have the regulatory framework to introduce PMACS. Both these regulators and MAHs could propose PMACS, if preferable. This study demonstrated Naglazyme (galsulfase) (BioMarin Pharmaceutical Japan K.K., Japan) as a case where PMACS or clinical surveillance programs like PMACS have been required in each region/country.
Limitations
This study targeted the reexamination reports issued in 2017–2019 to determine the implementation of PMACS after the PMDA was established, as mentioned in the ‘Materials and Methods’ section. The concept of safety measures has advanced in Japan since the establishment of the PMDA, including the introduction of the Risk Management Plan in 2013. 24 Therefore, the latest trends in the implementation of PMACS could be observed once further investigation is conducted after the relevant reexamination reports are published.
Footnotes
Author contributions
Both the authors made substantial contributions to designing the study, collecting data, examining the relevant information, and drafting and reviewing this paper. Dr Masamune has taken the final responsibility to submit this paper in this journal.
Conflict of interest statement
Hideyuki Kondo is a employee of Novartis Pharma K.K.
Ethical approval/patient consent
Not applicable, as this study did not recruit any patients or provide intervention to them. The study was conducted based on public data.
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.