
Abstract
A favorable benefit–risk profile remains an essential requirement for marketing authorization of medicinal drugs and devices. Furthermore, prior subjective, implicit and inconsistent ad hoc benefit–risk assessment methods have rightly evolved towards more systematic, explicit or “structured” approaches. Contemporary structured benefit–risk evaluation aims at providing an objective assessment of the benefit–risk profile of medicinal products and a higher transparency for decision making purposes. The use of a descriptive framework should be the preferred starting point for a structured benefit–risk assessment. In support of more precise assessments, quantitative and semi-quantitative methodologies have been developed and utilized to complement descriptive or qualitative frameworks in order to facilitate the structured evaluation of the benefit–risk profile of medicinal products. In addition, quantitative structured benefit–risk analysis allows integration of patient preference data. Collecting patient perspectives throughout the medical product development process has become increasingly important and key to the regulatory decision-making process. Both industry and regulatory authorities increasingly rely on descriptive structured benefit–risk evaluation and frameworks in drug, vaccine and device evaluation and comparison. Although varied qualitative methods are more commonplace, quantitative approaches have recently been emphasized. However, it is unclear how frequently these quantitative frameworks have been used by pharmaceutical companies to support submission dossiers for drug approvals or to respond to the health authorities’ requests. The objective of this study has been to identify and review, for the first time, currently available, published, structured, quantitative benefit–risk evaluations which may have informed health care professionals and/or payor as well as contributed to decision making purposes in the regulatory setting for drug, vaccine and/or device approval.
Plain language summary
The review of the benefits and the risks associated with a medicinal product is called benefit–risk assessment. One of the conditions for a medicinal product to receive marketing authorization is to demonstrate a positive benefit–risk balance in which the benefits outweigh the risks. In order to enhance the transparency and consistency in the assessment of benefit–risk balance, frameworks and quantitative methods have been developed for decision making purposes and regulatory approvals of medicinal products. This article considers published quantitative benefit–risk evaluations which may have informed health care professionals and/or payor as well as contributed to decision making purposes in the regulatory setting for drug, vaccine and/or device approval.
Keywords
Introduction
The evaluation of the benefit–risk balance is a key element across the entire life cycle of a medicinal product. Benefit–risk assessments support decision-making purposes during the development and the regulatory process of approval, as well as post-marketing follow-up and payor/health care provider selection. Typically, a qualitative description of evidence has been the standard approach to establish a drug’s benefit–risk profile.
Recently, assessment of benefit–risk profiles for medicinal products have shifted from an unstructured, subjective and inconsistent approach using disparate sources of data selected by different experts towards a more structured approach. 1 Structured benefit–risk evaluation provides a more objective assessment of the benefit–risk profile of medicinal products and a higher transparency for decision making purposes. Accordingly, varied structured frameworks and quantitative methodologies have been developed to make evaluation of benefit–risk profiles of medicines more comprehensive and consistent.
Structured benefit–risk assessment frameworks can be divided into two categories: (a) descriptive and (b) quantitative. These have been described elsewhere in systematic reviews of benefit–risk assessment methodologies.2,3
Descriptive frameworks include the Food and Drug Administration’s (FDA’s) benefit–risk framework, 4 PrOACT-URL (problem, objectives, alternatives, consequences, trade-off, uncertainly, risk tolerance, and linked decisions)5,6 and BRAT (Benefit–risk Action Team). 7 Each of these frameworks provides qualitative stepwise instructions for the benefit–risk evaluation. While they all allow for structured benefit–risk evaluations, none offers quantitative methods to integrate benefit and risk outcome measures or incorporate preference weights for benefit and risk criteria into the assessment.
In contrast, quantitative frameworks utilize systems 8 with varied scope and complexity, to provide explicit methods for balancing the benefits and risks. The Pharmacoepidemiological Research on Outcomes of Therapeutics by a European Consortium (PROTECT) has been a key contributor in providing frameworks and methodological recommendations through practical examples.2,3,9,10 However, what remains unclear is how frequently these quantitative frameworks have been used by pharmaceutical companies to support submission dossiers for drug approvals and their impact in regulatory decisions in the context of drug, vaccine and device approvals.
Multi-criteria decision analysis (MCDA)8,11 has been the most popular method for quantitative benefit–risk assessment studies. Incorporation of preference weights from different stakeholders, including patients, for key outcomes is an essential aspect of it. Patients are a key stakeholder to assess a drug’s acceptable risk and define the level of risk they are willing to accept to reach meaningful clinical benefits.
As such, integrating patient perspectives is recently becoming an important part of the regulatory decision-making process and pharmaceutical industries actively engage patients to collect their perspectives throughout the development and life cycle of medical products. 4
This article reviews available structured quantitative benefit–risk evaluations which have been published and may have informed health care professionals and/or payor as well as contributed to decision making purposes in the regulatory setting for drug, vaccine and/or device approval.
Structured benefit risk: descriptive and quantitative methods
A structured benefit–risk assessment should be initiated with a descriptive framework from which subsequent quantitative analysis may proceed. This is demonstrated in Figure 1 in the algorithm developed by PROTECT to define the steps that guide the choice of the appropriate benefit–risk methodology. If the conclusion of the qualitative benefit–risk assessment does not provide a clear picture regarding which medicinal product is preferred (question “is one alternative clearly most preferred?”), a quantitative approach should be undertaken.

Algorithm to define the appropriate benefit–risk methodology (based on the PROTECT recommendations), extracted from Hughes et al. 3
The benefit–risk assessment frameworks can be divided into two categories: (1) descriptive and (2) quantitative (Figure 2).
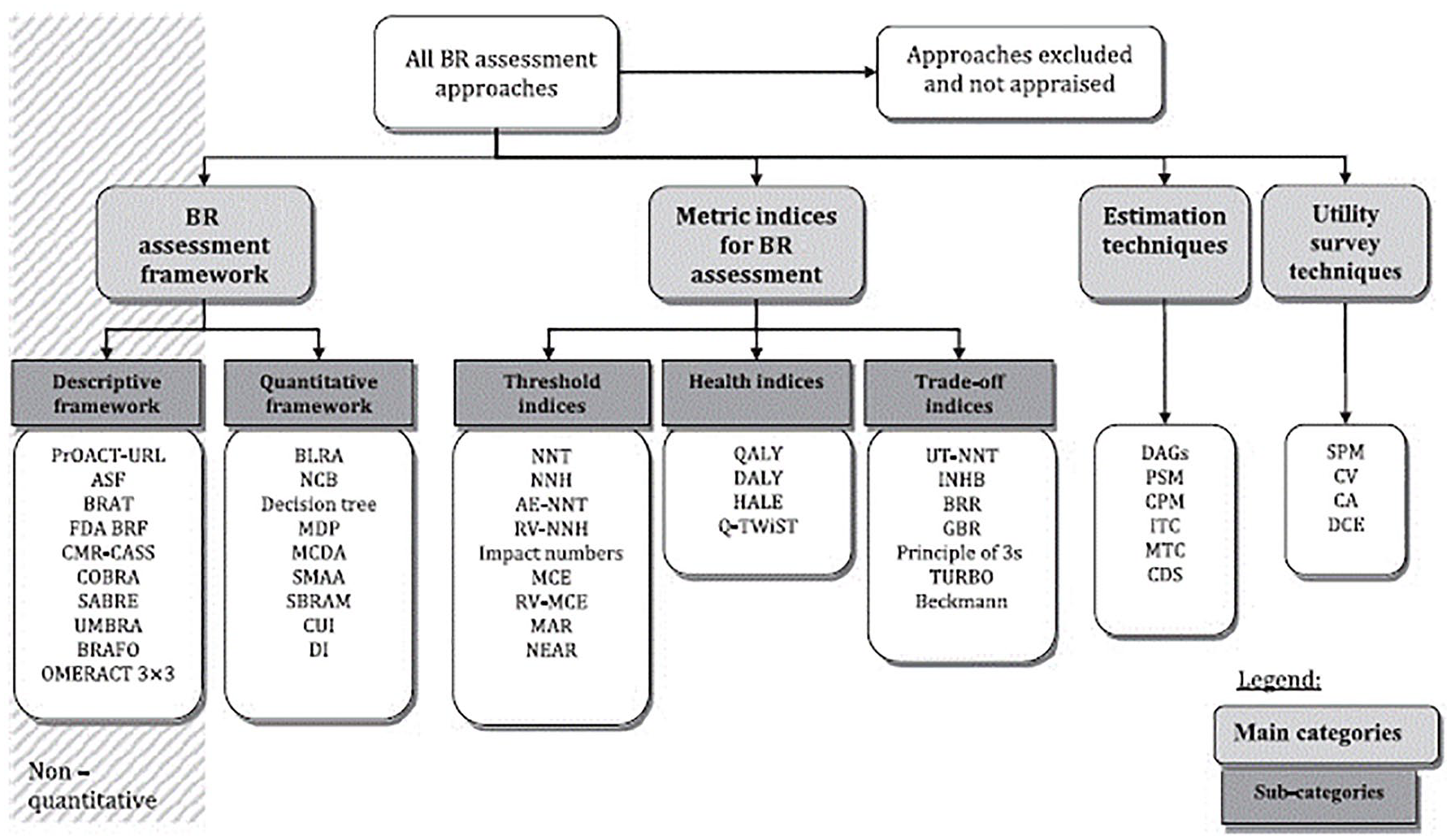
Overview of descriptive and quantitative frameworks from PROTECT, extracted from PROTECT website. 10
Descriptive frameworks
There are several descriptive frameworks and the commonly used are described below.
FDA benefit–risk framework
The FDA’s benefit–risk framework is a structured, qualitative approach focused on identifying and clearly communicating key issues, evidences and uncertainties for benefit–risk assessment and regulator decision-making.
As committed in the PDUFA V, the FDA has incorporated structured benefit–risk assessment in the human drug review process by integrating the benefit–risk framework into CDER (Center for Drug Evaluation and Research) and CBER (Center for Biologic Evaluation and Research) clinical review templates. Consequently, the benefit–risk framework is now publicly available and posted at drugs@FDA as part of the medical review for all original New Drug Applications after 2015. It is a valuable tool for explaining and communicating reasoning behind the FDA’s regulatory decisions in drug approvals, and to inform non-regulatory stakeholders for their own decision making in developing therapies, filing submission dossiers and speculating on similar FDA benefit–risk assessments in the future.
PrOACT-URL and BRAT frameworks
The descriptive frameworks PrOACT-URL and BRAT are useful guides for planning and executing benefit–risk assessments at any stage of a medicinal product’s life cycle. Each framework provides a structure for breaking down a benefit–risk problem into a stepwise evaluative process. The step progression for PrOACT-URL and BRAT are analogous, with slightly different process and decision matrixes.
These three descriptive frameworks, despite having their own specificities, share common features such as the logical soundness, practicality, consistency and transparency. They provide systematic approaches to identify most relevant benefits and risks and to define the rationale for each criterion to be included or not in the assessment. All three frameworks also use a decision context (indication, medical need, population, availability of other treatments, and perspective from which the decision is being made). Other common characteristics include the quality and interpretation of evidence, the transparency of prioritization and weighting of benefits and risks and the tracking of uncertainties. In fact, in addition to evaluating the strengths of the data submitted from randomized clinical trials and other data sources, the benefit–risk analysis must also take into account the uncertainties and limitations contained within this body of evidence (e.g. uncertainties due to limitations in study design, small sample size, etc.), as these will impact the overall conclusions. Finally, they also share the evaluation, the summary and the communication of relevant data and information via value trees or tables. 12 Despite allowing for structured benefit–risk evaluations, neither of them offers quantitative methods to integrate benefits and risks and to incorporate preference weight into the benefit–risk evaluation.
Quantitative methods
There are numerous quantitative methods (Figure 2), which vary by scope and complexity. Among the most popular methods, the multi-criteria decision analysis was identified by the European Medicines Agency (EMA) as a promising methodology for the quantitative benefit–risk assessment of medicines. 9 Multi-criteria decision analysis is frequently utilized for structured benefit–risk assessment studies. The steps that are needed to complete a multi-criteria decision analysis are the following: 12
Establishing the decision context (identify the medicinal product, therapeutic area and indications for which the assessment is to be done, defining the decision problem, what is to be decided and by whom);
Considering the alternatives (placebo, or competitive drug, or gold standard);
Selecting key benefits and risks to be represented in a value tree (identify and define the criteria for assessing the effect for the products of interest and for each alternative). [The key benefits are defined by favorable effects that contribute importantly to the overall benefit–risk evaluation and that are important for the patient (clinically important, relevant, intense or durable). The key risks are defined by unfavorable effects that contribute importantly to the overall benefit–risk evaluation. The selection is based on medical judgment (clinically important risks because of their severity, frequency, duration, toxicity, predictability, reversibility, or inability to be prevented). They may also include those that are considered for risk minimization activity beyond labeling.];
Relative weighting by establishing a measurement scale for each key benefit and risk and assess the relative importance of the scales. Several weighting methods exist, among which are discrete choice experiments and the swing weighting; they are described elsewhere; 13
Estimating the benefit–risk score (by combining the outcome measures of each criteria with corresponding weights) to describe how the alternatives perform for each of the criteria;
Displaying results (provide graphical displays);
Performing sensitivity analysis (by exploring effects of uncertainty on the benefit–risk balance);
Formulating the conclusion and recommendation.
The key advantage of the multi-criteria decision analysis model is application of relative weighting of benefits and risks by common value units in order to comprehensively and quantitatively assess products’ favorable and unfavorable effects. This involves integration of key stakeholder preference weights, such as the patient preferences. Multi-criteria decision analysis quantitative assessments based on different stakeholders may have complementary or contrasting conclusions, indicating potential disconnections among stakeholder opinions. Indeed, patients, physicians and regulators may have different views on the importance of one treatment outcome compared with another. Insight derived from such results contributes to overall perceived product value and possible opportunities for stakeholder engagement and education.
Quantitative structured benefit–risk: a key component for decision-making in the regulatory setting
Although multi-criteria decision analysis was identified by the EMA as a promising methodology for the quantitative benefit–risk assessment of medicines, 9 there is limited publicly available information regarding how frequently these methods are being used by pharmaceutical industries to support submission dossiers of drug approval or to respond to health authorities’ requests on the benefit–risk profile of medicinal products or devices. Thus, we searched in MEDLINE for published structured quantitative analysis in English up to 15 June 2020. The focus of this search was on the published studies that have used multi-criteria decision analysis to evaluate the benefit–risk profile of drugs or vaccines during the development stage or in the post-marketing settings. Articles focusing on the methodological aspects and/or providing guidance on the application of the multi-criteria decision analysis were not selected for this review. The following search terms were used: MCDA AND (benefit risk assessment OR risk benefit assessment OR BRA). Eligible articles were first selected based on their title and abstract, then based on the review of the full report. For each eligible study, we searched in CHMP, EMA, FDA websites and available reports to identify any evidence that the reported quantitative assessments where utilized in the regulatory process.
In total 117 hits were found through the search of databases, out of which nine publications were selected. These articles are listed in Table 1.
Studies using multi-criteria decision analysis (MCDA) quantitative method to evaluate the benefit–risk profile of drugs or vaccines during development or post-marketing stages.
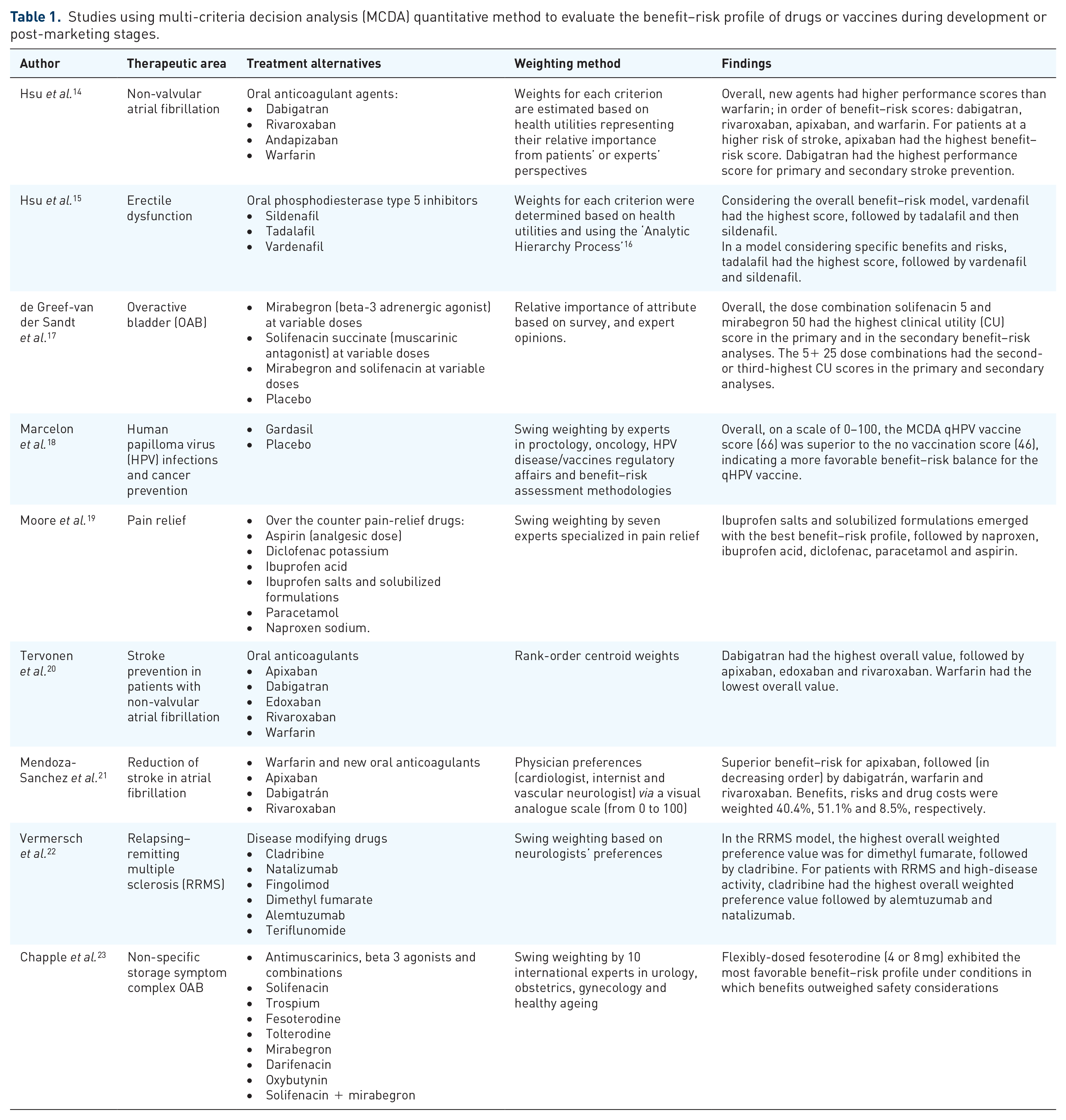
All of these papers had common objectives to compare the benefit–risk profile of several products using the quantitative, multi-criteria, decision analysis method and integrating data from several data sources of varied quality level. However, some specificities need to be highlighted. The study by Moore et al. 19 relied on the assessment of the benefit–risk balance of over the counter products. Most of the existing evidence derives from knowledge and clinical experience with prescription analgesics used at higher doses and for prolonged periods; thus, the challenge was to find a way to use these disparate data sources. Other studies used newly developed weighting methods14,15,20 or inclusion of criteria beyond clinical events, such as interaction with food, real world evidence data 20 or annual cost per person. 21 In the study by de Greef-van der Sandt et al., 17 multi-criteria decision analysis was used at the early stage of development (based on phase II data) in order to find the optimal dose of the drug combination to be further investigated in the phase III study.
Quantitative structured benefit–risk assessments are also valuable when used in the regulatory setting for product approval, or immediately post-approval, as provided in the following examples.
The first example was conducted by Marcelon et al. for the quadrivalent recombinant human papillomavirus (HPV; types 6, 11, 16 and 18) vaccine (qHPV) (Gardasil/Sigard). 18 This vaccine was first approved by the European Union on 20 September 2006 for the prevention of premalignant cervical, vulvar and vaginal lesions and cervical cancer due to HPV types 16 and 18; and of condyloma acuminata due to HPV types 6 and 11. In 2014, the marketing authorization holder resubmitted an application to extend the indication to include pre-malignant anal lesions and anal cancer. In order to support the submission dossier and help regulatory decision making for the indication extension, the marketing authorization holder conducted a structured qualitative and quantitative evaluation of the benefit–risk balance of Gardasil vaccination using two complementary methods. Both the PrOACT-URL framework and the multi-criteria decision analysis were used to estimate the overall benefit–risk balance of qHPV compared with “no vaccination” as an alternative option in the prevention of premalignant anal lesions and anal cancer. Based on this analysis, the marketing authorization holder concluded that the benefit–risk profile of qHPV vaccine was positive with respect to prevention of anal cancer, with impact on genital warts being the most important beneficial effect. Consistently, the Committee for Medicinal Products for Human Use (CHMP) considered the multi-criteria decision analysis conducted by the marketing authorization holder to be a tool of interest supporting and complementing the submitted follow-up data on duration of protection and safety profile from clinical trials and post-marketing observational studies. Based on the review of the submitted data, the extension of the indication to include prevention of premalignant anal lesions and anal cancer was accepted. The method and results of this analysis were also included in the CHMP assessment report 24 and were detailed in a publication.18,25
The second published example is the case of cladribine (MAVENCLAD®), a disease modifying drug indicated for the treatment of adult patients with highly active relapsing–remitting multiple sclerosis (RRMS). Given the large number of approved disease modifying drugs for the treatment of patients with RRMS and/or other forms of relapsing multiple sclerosis, balancing efficacy versus the risks associated with those treatments is essential to the selection of a treatment option for every patient with RRMS and particularly for those patients at increased risk of relapses and disability progression.
In 2016, the marketing authorization holder submitted a marketing application to the EMA for MAVENCLAD® (cladribine) for the indication “treatment of adult patients with highly active RRMS as a single disease modifying therapy”. During the scientific advisory meeting in 2017 the CHMP issued a positive opinion for granting a marketing authorization to MAVENCLAD. 26 In parallel to this marketing application, a multi-criteria decision analysis was applied to the benefit–risk assessment of cladribine and five other approved disease modifying drugs (alemtuzumab, dimethyl fumarate, fingolimod, natalizumab and teriflunomide). Five independent European neurologists participated in this analysis in 2015 to provide weights on the selected key benefits and key risks. 22 The results of the analysis, considering the expert judgment on the favorable and unfavorable effects of disease modifying drugs, concluded that cladribine is an important treatment alternative to consider both in patients with RRMS and in patients with RRMS with high disease activity (high disease activity is defined as two relapses in the previous year). 22 Furthermore, this result aligned with the conclusion drawn by the EMA during the European authorization application’s assessment, who considered the overall benefit–risk balance of cladribine as favorable in the treatment of adult patients with highly active relapsing multiple sclerosis.
Discussion
This study is the first to review examples of the application of published quantitative benefit–risk approaches in drug development. Based on our findings, it is interesting to note that although the structured benefit–risk methodologies have been widely discussed and presented in the regulatory landscape, there are surprisingly few published examples and limited evidence of quantitative structured benefit–risk applied in the regulatory setting. This could be related to limited use of these methods but is likely due to low transparency use of the results for confidential decision-making.
In addition, patient preferences are not always considered in quantitative evaluation. Instead, physician perspectives are generally used. This limitation was observed in both multi-criteria decision analyses of Gardasil and cladribine presented above. This suggests that, although there is a growing interest in incorporating patient preferences information within decisions, there is still a lack of input from the patients along the medical product life cycle. Quantitative structured benefit–risk methods provide an excellent opportunity to incorporate the patient voice in the decision-making process and this concept must see wider utilization.
Patient preferences in benefit–risk assessments have become increasingly important for the marketing authorization holders or applicants and for health authorities, not only when developing or approving a new medical product but also for all decision making throughout the product life cycle. 27 Indeed, patients have individual perspectives of the most important benefits and acceptable risks of their own treatment options. Because only patients live with their medical condition and impact on health and quality of life, preferences relating to benefit and risk tradeoffs may be different from the perspectives of regulators and health care providers. 28
An early example of patients’ voices being heard relates to the HIV epidemic in the 1980s, when patient perspectives were included in the drug approval process for antiviral medicines, which contributed to timely approval of the first protease inhibitor in 1995.
More recently, the first patient-preference study on weight-loss devices was designed to obtain quantitative patient-preference evidence to inform regulatory decision-making. It quantified the relative importance of safety and effectiveness and other attributes of weight loss devices to obese individuals. 29 The Center for Devices and Radiological Health (CDRH) partly used the results derived from this survey to determine that obese patients are willing to take more risks in exchange for weight loss and to support approval of the device in 2015, marking the first approval of a new device as a result of the CDRH’s Patient Preference Initiative. 28
Another example is taken from the oncology area. A stated preference study in a sample of patients with relapsed or refractory multiple myeloma from the cancer charity Myeloma UK was conducted. 30 The objectives of this study were to elicit the preferences of patients with multiple myeloma regarding the possible benefits and risks of cancer treatments and to illustrate how such data may be used to estimate patients’ acceptance of new treatments. This study demonstrated how quantitative preference statements from a large group of participants can be collected through an online survey and how such information may be used to explore the acceptability of specific treatments based on the attributes studied. Notably, this study also showed considerable heterogeneity with respect to the relative importance given to the risk criteria within this large group of participants.
This introduces another field of interest, which is the personalized benefit–risk assessments31–33 in which individual patient data are considered instead of consolidated data describing an “average” patient. For more information on this topic, a recent example was published for the oral antiplatelet vorapaxar by combining trial and real-world data to further personalize the treatment profiles. 33
There is a wide range of descriptive and quantitative methods that can be considered for benefit–risk assessment. Each of these methods has its specificities, its strengths and limitations but the main objective of these tools is to enhance the interactions between assessors and stakeholders and to increase the transparency of decisions across the product life cycle. A stepwise approach should be considered in order to guide the choice of the most appropriate benefit–risk methodology starting as a minimum with the structured descriptive framework. Structured quantitative benefit–risk frameworks and methods should be considered when needed. More specifically, these analyses should be considered for complex situations where several treatment options are available and when the descriptive framework does not clearly allow a definitive conclusion on the preferred medicinal products for a targeted indication or specific population of interest. The quantitative approach should integrate key stakeholder’s preferences, especially the patient preferences. The collection of patient preference weights for benefits and risks is, however, one of the challenges in quantitative benefit–risk methods. It requires quantitative patient preference data to be collected under controlled conditions to obtain valid and reliable preference data.
Based on a recent evaluation, 34 more than 20% of the submissions to FDA made between 2018 and 2019 did not include patient data, meaning that in one out of five submissions, patients are not yet considered as key stakeholders.
Conclusion
The use of a structured benefit–risk assessment with benefit–risk frameworks and quantitative methods, as well as the integration of patient preferences at the different stages of the product lifecycle, is crucial. It offers added value to ensure better transparency and consistency in decision-making processes. Quantitative benefit–risk evaluation should be used for complex situations as a complementary approach to assess the benefit–risk of a product. Engaging patients and collecting their perspectives throughout the medical product development process is gaining increasing recognition and becoming key in the regulatory decision-making process. Results from quantitative patient preferences studies are very important to gather patient views and to study heterogeneity in preferences in a systematic and structured way.
Despite the multiple public and private initiatives in developing different benefit–risk frameworks and tools, there is still a lack of a clear guidance and understanding on how regulatory bodies expect structured benefit–risk to be conducted, on how to integrate patients’ preferences and when to use quantitative methods. 35 Regulatory bodies should continue to collaborate with multiple stakeholders, including academia, industry, health-care professionals, patients and others to strengthen regulatory science and to address many critical knowledge gaps. 36 More research is warranted to address the inherent limitations and concerns related to the use of quantitative methods in benefit–risk assessments.
Footnotes
Conflict of interest statement
Marie-Laure Kürzinger, Ludivine Douarin, Ievgeniia Uzun, Chantal El-Haddad, William Hurst, Stéphanie Tcherny-Lessenot and Juhaeri Juhaeri are employees of Sanofi, a pharmaceutical company that manufactures various drugs and biologics. They did not receive a specific grant for this manuscript but salary from Sanofi.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.