
Abstract
The development of biosimilars is growing rapidly, especially in Europe. They are a cost-effective alternative to original biological medicines and can help improve patient access to these therapies.
The European Medicines Agency (EMA) has been the first to issue scientific guidelines related to regulatory requirements for the approval of biosimilars. These guidelines have been being updated in line with advances in analytical techniques and growing experience in the clinical use of these drugs.
Given the complex nature of biological medicines, they pose a greater potential risk of immunogenicity than nonbiological medicines, and hence warrant special consideration. The risk management plan for biopharmaceuticals (innovator and biosimilar drugs) should be based on strengthening ongoing pharmacovigilance activities, especially in the post-approval period.
This paper addresses regulatory issues related to the approval of biosimilars in Europe associated with safety considerations linked to the development and use of these medicines. We also discuss the issues of immunogenicity, interchangeability and traceability of biological medicines.
Keywords
Introduction
Biological medicines represent a major advance in the treatment of serious pathological conditions such as cancer and neurodegenerative and autoimmune diseases. 1 Unlike small-molecule drugs which have a perfectly defined molecular structure and are chemically synthesized, biological medicines are produced by biotechnological methods. 2 Given that they are obtained from live organisms, no two biological drug substances are exactly the same. After 10 years from the approval of the original biologics, companies other than the originator company may obtain a European Union (EU) license for a biosimilar medicine.
The European Medicines Agency (EMA) was a pioneer internationally in establishing the regulatory basis for the development of biosimilars. This agency defines a biosimilar medicine as a biological medicinal product that contains a version of the active substance of an already authorized original biological medicinal product (reference medicinal product) in the European Economic Area (EEA). 3 Since the approval of the first biosimilar of somatotropin in 2006, the EMA has issued several scientific guidelines regarding the regulatory requirements for the approval of biosimilar drugs. These guidelines have been being updated in line with advances in analytical techniques and growing experience in the clinical use of these drugs. 3 A total of 39 biosimilars have been approved through the EMA for use in the EU as of March 2018.
One of the greatest concerns about biological medicines is related to their safety. The knowledge of risks and benefits of the product at the time of approval is liable to change over time through expanded use, in terms of patient characteristics and the number of patients exposed. For this reason, pharmacovigilance should be ongoing for these products. 4
This paper addresses regulatory issues related to the approval of biosimilars in Europe associated with safety considerations linked to the development and use of these medicines.
Biosimilar development
The development of biosimilars is based on the demonstration of biosimilarity, through a comparability exercise (‘comprehensive head-to-head comparison of the biosimilar with the reference medicine to show high similarity in chemical structure, biological function, efficacy, safety and immunogenicity’ as described by the EMA). 5 The development of a new biological medicine is expensive, considering the costs for completion of the steps from analysis to preclinical and large clinical studies necessary to demonstrate their efficacy and safety. In contrast, the development of biosimilars is mainly based on quality studies, to demonstrate their biosimilarity to the reference product.
Figure 1 outlines the various steps in the development of a biosimilar. Few data on clinical efficacy and safety are required for the approval of a biosimilar compared with that of the original reference product, since the objective is not to demonstrate its efficacy and safety, as these have already been proven for the originator. Nevertheless, once a biosimilar has been approved, there should be ongoing analysis of its clinical safety, as occurs for innovator products. 6
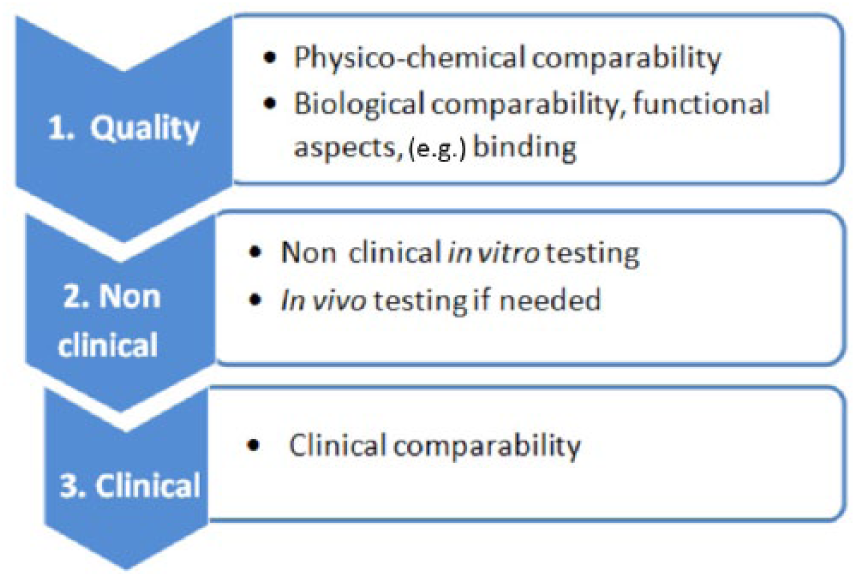
Biosimilar development: steps in the comparability exercise.
Safety of biologicals
The safety profile of biological medicines depends on their pharmacological properties, including immunological properties of the drug, as well as the properties of the excipient and any process-related impurities, and the susceptibility of the patient, that is, medicine-induced allergic reactions, autoimmunity, etc. 7
Immunogenicity
Given the complex nature of biological medicines, they pose a greater potential risk of immunogenicity than nonbiological medicines, and hence warrant special consideration.8,9 Immune responses may be responsible for adverse reactions, which may be acute and delayed. Acute reactions can cause severe hypotension, bronchospasm, laryngeal or pharyngeal oedema, wheezing or urticaria, among other symptoms, while delayed reactions may give rise to other symptoms, such as myalgia, arthralgia, skin rash, or pruritus. 10
Although generally immunological reactions to biological medicines have no clinical consequences, in some cases, such reactions can be serious, causing hypersensitivity, infusion reactions, anaphylaxis, and loss of efficacy, and may even be life threatening. 11 Therefore, one of the most important issues with regard to the safety of biological medicines, including biosimilars, is their immunogenic potential.
The clinical consequences of immunogenicity include the partial or total loss of efficacy of the drug involved, and this can be due to the production of antidrug antibodies (ADAs), changes in pharmacokinetics, and potential induction of cross-reactivity to endogenous proteins, among other factors. Therefore, one of the tests that should be carried out during product development is the assessment of immunogenicity. 10
Various different factors, including genetic ones, influence the possibility of the induction of an immune response against therapeutic proteins: patient characteristics and comorbidities, the drug administration regimen, product characteristics and changes in the manufacturing process, among others. Specifically, the immune response to biological agents may vary with patient age. The immune system matures with age, and hence the immune response to biological medicines may differ in children. Similarly, the immune response may be altered among elderly people. 12
Another factor that may determine the triggering of an unwanted immune response is the patient’s health status. That is, patients with an activated immune system (e.g. due to allergies, autoimmune conditions, or chronic infections) may be more prone to developing immune reactions against biological medicines. 10 The concomitant administration of other pharmacological treatments may reduce or increase the risk of immunogenicity to biologics. For example, a weakening of the immune response is observed when they are administered in combination with immunosuppressants. 13
Additionally, the dose, dosing regimen and route of administration may all have an effect on the immune response. Generally, short-term treatments are less likely to trigger a dangerous immune response than long-term treatments, and intravenous administration is associated with less immunogenicity than subcutaneous or intramuscular routes. 10
Lastly, the immunogenicity of biologics depends on characteristics of the product itself (e.g. post-translational modifications, changes in the structure of the molecule during inappropriate storage or transport), presence of impurities related to the product or process (for example, host cell proteins), the excipients, the formulation and the container. 14
Analysis of comparative immunogenicity
A comparability exercise involves analysis of the immunogenicity of a biosimilar and of the reference medicine. The purpose of this type of analysis is to assess the immune response in both the short term (e.g. infusion-related reactions) and the long term. In addition, the tests generally detect antibodies against all epitopes of both biosimilar and reference product molecules. 15
In the case of monoclonal antibodies (mAbs), attention should be paid to some particular issues, since the risks of immunogenicity may have consequences that are particularly important in these drugs. For example, a change in the glycosylation patterns may alter the immunogenic properties of the biological product. This may also occur with the use of different expression systems, potentially resulting in a higher risk of immunogenicity than that observed when using the usual systems. 16
If antibodies are produced against mAbs, so-called ADAs, the clinical effect depends on the antibody binding site, the affinity of the antibody for the mAb and the amount of antibody developed. In some cases, the development of antibodies may have no significant clinical effects, but in others, efficacy may be reduced or there may be adverse effects. Specifically, ADAs may neutralize mAbs and reduce their efficacy or give rise to adverse effects such as infusion-related reactions and immune complex formation. 16
Pharmacovigilance
Any medicine has the potential to produce adverse reactions, with varying levels of severity and frequency. Nevertheless, not all adverse reactions are found before the approval of a new drug, some of them only being discovered during post-marketing use. 10
As part of the marketing authorization process for biological medicines, like that for other drugs, the marketing authorization applicant must submit a pharmacovigilance plan as part of a risk management plan (RMP) to the relevant authorities in accordance with European regulations. Applicants seeking the approval of a biosimilar should also submit an RMP, as would be required for originals. Monitoring of the safety of biosimilars is subject to the same requirements as those that apply to all biological medicines. 4
The purpose of an RMP is to document the risk management system necessary to identify, characterise and minimize a medicinal product’s important risks; and to achieve this, it should: 6
- identify the safety profile of the medicine, paying special attention to important identified and important potential risks (the so-called ‘safety specification’);
- outline a plan for pharmacovigilance activities, seeking to characterise and quantify clinically relevant risks, and to identify new adverse reactions (the so-called ‘pharmacovigilance plan’);
- outline a set of risk minimization measures and describe their implementation and evaluation (the so-called ‘risk minimization plan’).
An RMP for a biosimilar must consider the risks observed during the use of the reference product as well as potential risks. It has also to indicate how to manage the risks throughout the product’s post-marketing life. One of the specific factors to recognise in the RMP is the risk of immunogenicity. In the case that the RMP includes immunogenicity in the safety specification, routine risk minimization activities may be required. The description of these activities may consist of, among other measures, the measurement of ADAs and trough levels (minimum levels) as well as how to act in the event of ADA development and related adverse reactions.
The RMP should focus on the important identified risks that are likely to have an impact on the benefit–risk balance of the product. In general, whenever the RMP of the reference product is updated, the biosimilar RMP should also be updated in the light of the new information. Any safety monitoring of the reference product or product class should also be included in the biosimilar pharmacovigilance plan, or its omission justified.
The safety specification component of the RMP includes a summary of the important identified and potential risks of the medicine, as well as missing information. 4 Additionally, it should specify the populations potentially at risk. The safety specification provides the basis of the pharmacovigilance plan and the risk minimization plan.
The RMP is a dynamic document that should be updated throughout the life cycle of the corresponding medicine. The marketing authorization holder should provide a summary of the RMP for the medicinal product, which is published on the EMA website, together with the other documents of the European Public Assessment Report (EPAR) of that medicinal product. The EPAR gathers the product information and is a set of documents describing the evaluation of a medicine authorized via the centralized procedure.
The regulatory control of RMPs for medicines authorized centrally is carried out by the Pharmacovigilance Risk Assessment Committee (PRAC) in Europe. Nevertheless, the EMA may also consult healthcare professionals and patients during the assessment of RMPs to propose risk minimization measures.
There are various different systems in place to monitor adverse drug reactions, both during clinical trials and during post-marketing use. They are described in detail in the EU pharmacovigilance legislation which came into force in July 2012. 17 Adverse reaction reports may arise from various sources including clinical trials, epidemiological studies, and spontaneous reports. In the EEA, suspected adverse drug reactions are reported through EudraVigilance, an advanced pharmacovigilance system for the monitoring, evaluation and prevention of drug-related adverse reactions. 18 At the end of 2017, EMA launched a new and improved version of EudraVigilance, that facilitates the reporting and analysis of suspected adverse reactions related to medicines and helps to support stronger safety monitoring. An overview of the current European pharmacovigilance system is available at the European Commission webpage. 19
With regard to pharmacovigilance, the marketing authorization holder is obliged to: 6
- Have an appropriate risk management system
- Ensure a critical review of the safety profile of the product, following its use in clinical practice. The marketing authorization holder must examine the pharmacovigilance data to assess whether there are new risks, whether the risks have changed and whether there are changes in the benefit–risk balance of the medicine. Critical review of a product’s safety profile is an ongoing activity, and the findings should be included in the information sent within the periodic safety update reports.
Interchangeability
In clinical settings, there is the option to exchange a reference product for a biosimilar (or vice versa) or replace one biosimilar with another. This practice is adopted when the two products are expected to have the same clinical effect. The ability to do this is what it is known as interchangeability.
If the prescriber decides to exchange one medicine for another with the same therapeutic purpose, this is known as switching. On the other hand, the concept of automatic substitution refers to the practice of dispensing one medicine instead of another equivalent medicine at pharmacy level without the knowledge of the prescribing physician.
In general, automatic substitution is not appropriate for biological medicines, including biosimilars. The approval of a biosimilar by the EMA does not include recommendations on whether the biosimilar is interchangeable with the reference medicine. Decisions on whether to allow interchangeable use and substitution of the reference biological medicine and the biosimilar are taken within the Member State.11,20
Traceability
When a medicine is suspected to cause an adverse reaction, in particular in the case of a biological medicine, it is essential to identify the product involved with the brand name, or international nonproprietary name (INN) and name of the marketing authorization holder, and batch number of the product administered, which should be clearly recorded in the patient file. In this case, the health professional must report the suspected adverse reaction to the national pharmacovigilance network through the completion of a report form. For this purpose, it is recommended that biological medicines are prescribed by brand name and not by INN. 21
Another approach that may contribute to the accurate recording in patient files is the use of bar code-scanning technology to unambiguously identify the product actually administered or dispensed to the patient. This may help to increase patient safety, reduce medication errors, and ensure successful adverse reaction reporting.
Reporting adverse reactions
Health care professionals must appropriately report safety signals of biological medicines and biosimilars. Reporting of a suspected adverse reaction must meet the same requirements for biological and nonbiological medicines. As stated above, the accurate identification of the biological product in the suspected adverse drug reaction report is essential, indicating the brand and batch number. Similarly, it is important that healthcare providers give patients and carers details of the brand and batch number of the biological administered or dispensed. 22
Given that immunogenicity is a specific source of concerns for biologicals, the related information must be provided systematically. In the event of a prior exposure to the same or cross-immunogenic products, this should be stated in safety communication reports.
It is important that patients and healthcare professionals are aware that adverse reactions may occur even if a medicine has previously been well tolerated. Such reactions may be due to delayed onset effects, but also due to variability in the drug production process. Given this, it is essential to report suspected adverse reactions occurring even after long-term use of a product. Further, we should provide information to users of biological medicines in order to avoid errors in their management, for example, on how to maintain the cold chain during delivery, storage, and handling. It should be remembered that biological medicines should be stored under refrigerated conditions, generally between 2°C and 8°C, as exposure to higher temperatures may result in denaturation of the constituent proteins. This could cause the product to lose its activity or become deactivated (irreversibly).
Additional monitoring
Once on the market, all drugs must be appropriately monitored, to check their efficacy and safety under real clinical conditions. Biological medicines are subject to so-called ‘additional monitoring’ and are included on a list of medicines under ‘additional monitoring.’ This concept concerns medicines authorized in the EU that are being monitored particularly closely by regulatory authorities because, for example, they contain a new active substance or limited data are available on their clinical use. These medicines subject to ‘additional monitoring’ are intensively monitored during the first few years after marketing authorization and are labelled with a black triangle (▼) on their package insert together with the sentence: ‘This medicinal product is subject to additional monitoring.’ 23
The concept of additional monitoring serves to encourage healthcare professionals and users to report suspected adverse drug reactions of new drugs. If a biological medicine (or biosimilar) is subject to ‘additional monitoring,’ it does not necessarily imply that there are particular safety concerns.
Extrapolation of indications
In the case of innovator medicines, efficacy and safety have to be demonstrated separately for each indication. In contrast, confirmatory clinical trials with the biosimilar are usually not required for all indications approved for the reference medicine. Once biosimilarity is proven, extrapolation of efficacy and safety data to other indications is always based on robust comparability studies. 15
Nevertheless, the extrapolation of indications varies between regulatory agencies. For example, the EMA approved the infliximab biosimilar for all the indications of the reference product (Remicade®), despite clinical trials only being carried out for rheumatoid arthritis and psoriasis. The data generated for these indications were extrapolated to the other indications of Remicade®, such as ulcerative colitis and Crohn’s disease. On the other hand, Health Canada initially did not support extrapolation of the clinical data to these other conditions, namely, Crohn’s disease and ulcerative colitis. This agency indicated that differences observed in certain in vitro functional assays, such as antibody-dependent cellular cytotoxicity (ADCC), between the biosimilar and the originator, were considered of potential relevance to the mechanism of action in inflammatory bowel disease. Nevertheless, Health Canada subsequently approved full extrapolation after additional data were provided by the sponsor. Prescribing physicians are advised to confirm that biosimilars are authorized for the target indication.24,25
The extrapolation of safety data can be justified if it has been proven that the biosimilar safety profile is comparable in a therapeutic indication/patient population that is sufficiently sensitive to differences in immunogenicity. 10 In this case, if the comparability of a biosimilar has been demonstrated, in terms of structure, functionality, pharmacokinetics, pharmacodynamics and efficacy, it can be anticipated that the adverse reactions of a biosimilar related to its pharmacological effect will be the same as and occur with a similar frequency to those of the reference product.
Utilisation of biosimilars
Given the high costs of biological therapies, biosimilars represent a cost-effective alternative to original biologicals and can help improve patient access to these treatments. 26 Additionally, biosimilar medicines stimulate price competition typically resulting in lower overall prices for a given product class, and thereby contribute to the sustainability of health systems. This has resulted in an increased utilisation of the class as a whole (biosimilar plus innovator drugs) in recent years, especially for some biopharmaceutical products, such as filgrastim.
As biosimilars are becoming more widely used and their quality is proven, they are earning the trust of clinicians. It is therefore essential to provide more information both on regulatory issues related to biosimilars and scientific matters to guide the medical decision making process. Training activities on the quality, safety, efficacy, extrapolation, and interchangeability of these drugs are recommended for both health professionals and patients. The EMA has published a guide that contains objective information for health professionals that could help in this task. 5 Such efforts would spread awareness and knowledge about biosimilars and help increase the trust in these biopharmaceuticals among all stakeholders.
Conclusion
The experience acquired since the introduction of biosimilars approved by the EMA has helped to strengthen our trust in them, with growing evidence that they can be used safely and effectively in their approved indications. In coming years, there is expected to be an avalanche of new biosimilars coming onto the market as the patents of innovator products expire.
Given the complex nature of biological medicines, they pose a greater potential risk of immunogenicity than nonbiological medicines, and hence warrant special consideration. Though generally immunological reactions to biologicals have no clinical consequences, in some cases, such reactions may be serious including hypersensitivity, infusion reactions, anaphylaxis, and loss of efficacy.
Applicants seeking the approval of a biosimilar must submit an RMP, as for original biological medicines. Further, the safety monitoring of biosimilars follows the same requirements that apply to all biological medicines.
Decisions on whether to allow interchangeable use and substitution of the reference biological medicine and the biosimilar is taken at national level. If an adverse reaction against a biological medicine is suspected, proper identification of the product is essential, providing details of the brand (or INN and name of the marketing authorization holder) and batch number of the product administered.
Training activities on issues such as quality, safety, efficacy, extrapolation, and interchangeability of these drugs should be run for both health professionals and patients.
Footnotes
Funding
The authors received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The authors declare that there is no conflict of interest.