
Abstract
Background:
Adverse drug reactions (ADRs) are common in older adults and frequently have serious clinical and economic consequences. This study was conducted as a feasibility study for a randomized control trial (RCT) that will investigate the efficacy of a software engine to optimize medications and reduce incident (in-hospital) ADRs. This study’s objectives were to (i) establish current incident ADR rates across the six sites participating in the forthcoming RCT and (ii) assess whether incident ADRs are predictable.
Methods:
This was a multicentre, prospective observational study involving six European hospitals. Adults aged ⩾ 65 years, hospitalized with an acute illness and on pharmacological treatment for three or more conditions were eligible for inclusion. Adverse events (AEs) were captured using a trigger list of 12 common ADRs. An AE was deemed an ADR when its association with an administered drug was adjudicated as being probable/certain, according to the World Health Organization Uppsala Monitoring Centre causality assessment. The proportion of patients experiencing at least one, probable/certain, incident ADR within 14 days of enrolment/discharge was recorded.
Results:
A total of 644 patients were recruited, evenly split by sex and overwhelmingly of White ethnicity. Over 80% of admissions were medical. The median number of chronic conditions was five (interquartile range 4–6), with eight or more conditions present in approximately 10%. The mean number of prescribed medications was 9.9 (standard deviation 3.8), which correlated strongly with the number of conditions (r = 0.54, p < 0.0001). A total of 732 AEs were recorded in 382 patients, of which 363 were incident. The majority of events were classified as probably or possibly drug related, with heterogeneity across sites (χ2 = 88.567, df = 20, p value < 0.001). Out of 644 patients, 139 (21.6%; 95% confidence interval 18.5–25.0%) experienced an ADR. Serum electrolyte abnormalities were the most common ADR. The ADRROP (ADR Risk in Older People) and GerontoNet ADR risk scales correctly predicted ADR occurrence in 61% and 60% of patients, respectively.
Conclusion:
This feasibility study established the rates of incident ADRs across the six study sites. The ADR predictive power of ADRROP and GerontoNet ADR risk scales were limited in this population.
Keywords
Background
Adverse drug reactions (ADRs) are a common cause of hospitalization and occur with increasing frequency in hospital as patients age. Research to date has focused largely on ADRs as a cause of hospitalization, and less on incident (in-hospital) ADRs. A recent meta-analysis reported that 6.3% of all hospital admissions are the direct result of ADRs, with an increased average attributable incidence of 10.7% seen in geriatric patients. 1 In other studies, focused exclusively on older adults, ADR prevalence rates as high as 26% have been reported. 2 Although less is known about ADRs that occur during hospitalization, a recent meta-analysis reported that 10.9% of all hospitalized adults experience an ADR, with 2.1% reported as serious. 3 A recent study in Ireland, focusing only on older adults, reported an incident ADR rate as high as 21% among unselected older patients hospitalized in a large tertiary referral centre with an acute illness. 4
The diagnosis of ADRs in geriatric patients can be challenging. Older people are a heterogeneous population, with high levels of multimorbidity and polypharmacy. Therefore, the ADR risk varies considerably between different geriatric patient groups. For example, nursing home residents are highly susceptible to medication related morbidity. 5 In addition, older adults experiencing ADRs often present with nonspecific symptoms such as cognitive decline, recurrent falls and reduced mobility, such that it can be difficult to discern whether medications have been implicated or not. ADRs have major clinical and economic consequences. They prolong hospital stay, 6 increase resource utilization, 7 can be fatal 8 and are costly. 9 Approximately one in two ADRs are thought to be preventable, 10 therefore there is major potential to avoid the associated morbidity, mortality and financial burden that accompany them. To date, few interventions have proven to be effective. Comprehensive geriatric assessment (CGA) reduces inappropriate prescribing (IP) and has the potential to reduce ADR incidence, 11 but this is resource intensive and time consuming. The application of STOPP (Screening Tool of Older Persons’ Prescriptions) and START (Screening Tool to Alert doctors to the Right Treatment) criteria by an experienced physician has recently been shown to be effective at reducing in hospital ADRs, with a difference in ADR rates of almost 10%, that is, an ADR rate of 21.0% in the control group versus 11.7% in the intervention group. 4
This study was conducted as a feasibility study for a forthcoming randomized control trial (RCT) for a large European Commission Seventh Framework Programme funded research consortium. This consortium will investigate whether a software assisted intervention advising on medication appropriateness [Software ENgine for the Assessment and optimization of drug and nondrug Therapies in Older peRsons (SENATOR)] will reduce incident ADRs among older patients who are hospitalized with acute illness. 12 This study was undertaken to (i) establish the incident ADR rates across the six international centres participating in the forthcoming RCT and (ii) assess whether incident ADRs can be predicted using the established GerontoNet ADR risk scale 13 and the newly developed ADRROP (ADR Risk in Older People) risk score. For the purpose of this study, ADRs were classified as incident if they occurred after enrolment into the study.
Methods
Study design and setting
This prospective observational study was conducted across six European hospitals over an 18-month period (February 2014–August 2015). The six centres involved were Cork University Hospital (Cork, Ireland), Universitair Ziekenhuis Gent (Ghent, Belgium), Hospital Universitario Ramón y Cajal (Madrid, Spain), Azienda Ospedali Riuniti (Ancona, Italy), Landspitali University Hospital Reykjavik (Reykjavik, Iceland) and Aberdeen Royal Infirmary (Aberdeen, Scotland).
Patient eligibility
Eligibility criteria in the feasibility study mirrored those of the proposed RCT (Supplementary information Table 1). Patients aged 65 years or older were eligible for inclusion if they were admitted as an emergency with an acute illness. Patients admitted under certain specialties, namely geriatric medicine, clinical pharmacology, palliative medicine, clinical oncology and haematology were excluded, as it was felt these specialties would dilute the impact of the intervention in the RCT by applying a substantial portion of the proposed intervention as part of routine clinical practice. For similar reasons, solid organ transplant recipients and those admitted to intensive care units were also excluded. Patients had to be recruited within 72 h of arrival to hospital, have an anticipated length of stay of >48 h, and require regular medications for at least three conditions. Participants also had to have a life expectancy of >3 months.
Outcomes
The primary endpoint of this study was the proportion of patients experiencing one or more nontrivial, probable or certain, incident ADRs within 14 days of enrolment or prior to discharge, whichever came first. For the purpose of this study, we applied the World Health Organization (WHO) definition of an ADR, that is, ‘a response to a drug which is noxious and unintended and which occurs at doses used in man for the prophylaxis, diagnosis or therapy of disease, or for the modification of physiological function.’ 14 Events were classified as prevalent if they occurred prior to enrolment and as incident if they occurred after enrolment. ADRs were viewed as discrete events, for example, if two drug-induced falls occurred, this was classified as two ADR events.
Adverse event ascertainment
Potential ADRs were referred to the Potential Endpoint Adjudication Committee (PEPAC) for review. The PEPAC consisted of the six-site primary investigators (PIs); all academic consultant physicians in geriatric medicine or clinical pharmacology with expertise in geriatric pharmacotherapy. To limit potential bias arising from the selective reporting of events to the PEPAC, a trigger list of the 12 most common ADRs, identified in earlier studies, was used. All such trigger list events were referred for review (Table 1). The trigger list was based on an earlier observational study 15 in which the clinical manifestations of the most common ADRs were defined (see also online Supplementary data). In this prospective observational study, ADRs were defined rigorously according to the World Health Organization Uppsala Monitoring Centre (WHO-UMC) causality criteria. Two physicians jointly reviewed all putative ADRs and only included those where there was consensus between them that a probable/certain ADR had occurred. The 10 most common ADRs reported were (i) acute kidney injury, (ii) electrolyte disturbance, (iii) falls, (iv) delirium, (v) constipation, (vi) orthostatic hypotension, (vii) dyspepsia, (viii) bleeding, (ix) diarrhoea and (x) symptomatic bradycardia. These ADRs formed the basis of the trigger list of potential ADRs, that is, adverse events (AEs) requiring further investigation. This trigger list was subsequently applied in a prospective randomized trial to identify ADRs. 4 The SENATOR consortium discussed the potential use of this trigger list and agreed it should be used for the feasibility study and RCT. Two additional AEs, on the basis of clinical experience, were proposed for inclusion, that is, gait disturbance and symptomatic hypoglycaemia. Their inclusion was agreed by consensus. The result was a 12-event trigger list designed to assist in unbiased reporting of ADRs. It was acknowledged that not all potential ADRs were included on the 12-point trigger list (for example, anaphylaxis). Therefore a generic nonprespecified ADR electronic evidence form was supplied to capture these AEs. As per the pre-specified events, a primary researcher (PR) could record these events and refer them for adjudication by the PEPAC.
Adverse event (AE) trigger list, definitions and total number reported in the study.
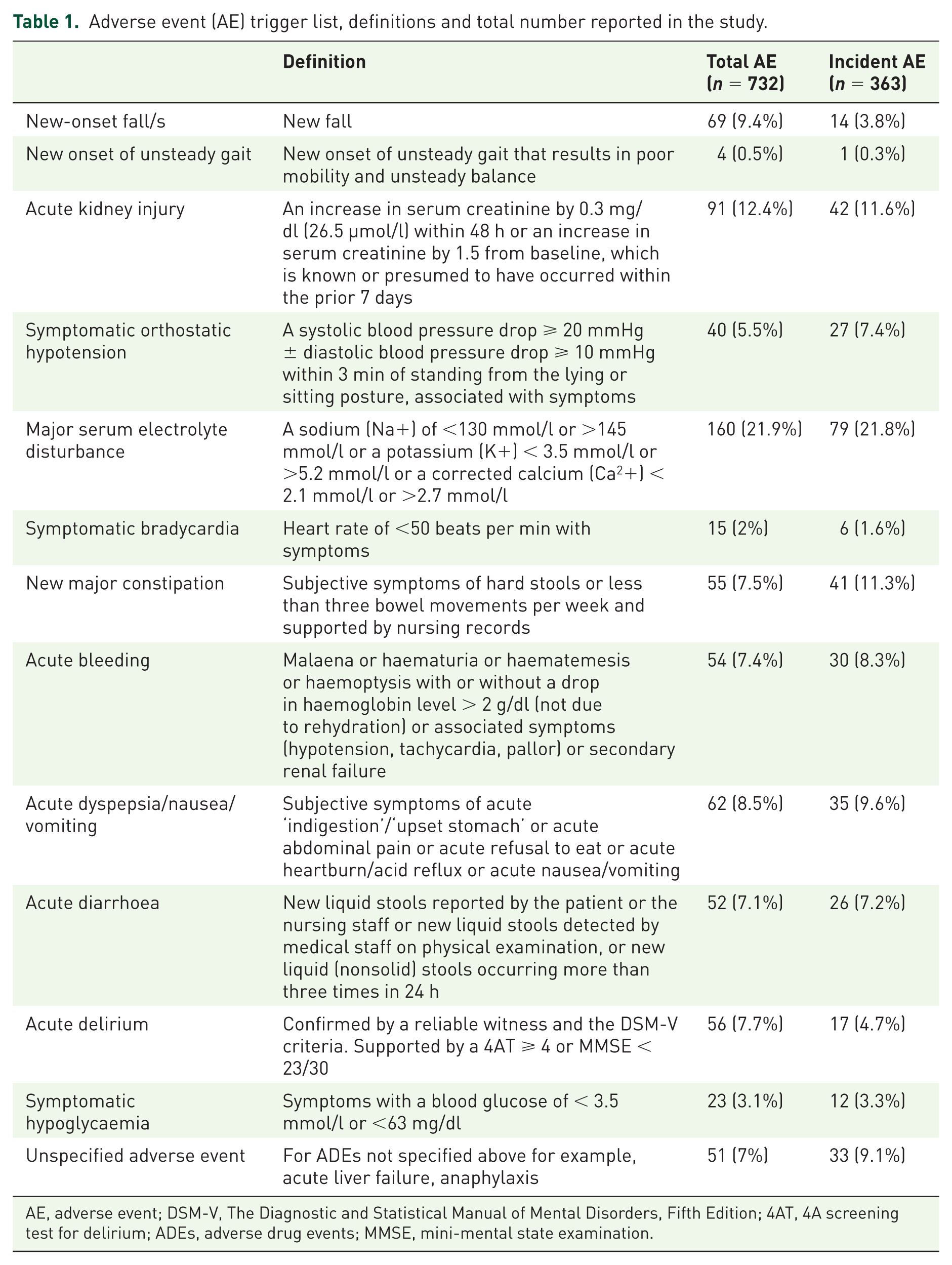
AE, adverse event; DSM-V, The Diagnostic and Statistical Manual of Mental Disorders, Fifth Edition; 4AT, 4A screening test for delirium; ADEs, adverse drug events; MMSE, mini-mental state examination.
This trigger list was used by the PR at two time points: (i) the point of recruitment at which time, prevalent ADRs were assessed in a prospective manner, and (ii) at day 14 postenrolment or at discharge (whichever came first), at which time, incident ADRs were assessed in a retrospective manner. At each time, they assessed whether any of the 12 AEs had occurred and if so, investigated further as to whether a drug was implicated. Thus, all prevalent and incident AEs, whether drug related or not, were recorded and subsequently reviewed to assess the causative role of current medications. ADRs were assessed retrospectively at day 14 or at discharge to avoid the possibility of PRs influencing ADR rates. Prospective assessments would have required regular contact between the PR, study patients and the medical team, and could have inadvertently increased medical teams’ awareness of ADRs. Prospective assessments could also have put PRs in a difficult position, for example, if a PR noticed a prescribing error that had the potential to cause an ADR, they would ethically be obliged to inform the medical team and this would influence the overall ADR rate. The main limitation of retrospective ADR ascertainment, was the potential to miss an ADR as ADRs are often not documented by medical staff in patients’ medical records. Thus, all incident ADRs were assessed by a locally trained PRs. PRs were encouraged to use all available data, that is, medical notes, nursing notes, laboratory records, medical prescriptions, allied health professional records, as well as imaging and additional tests. They were also encouraged to seek assistance from their site PI, if required. All AEs, along with any potentially implicated medications were recorded on a standardized electronic evidence form for review by the PEPAC. The level of causality was determined using the WHO-UMC criteria. 15 Each event, once recognized as potentially relating to a drug (possible, probable or certain as per the WHO-UMC criteria), had a level of severity (trivial, mild, moderate, severe) assigned according to a modified Hartwig and Siegel Severity Assessment scale. 16 Those that were adjudicated by the PEPAC to be probable or certain ADRs met the primary endpoint.
Consent
Patients provided written formal consent to participate in the study. In circumstances where patients were unable to give consent due to diminished decision-making ability (e.g. severe dementia or delirium), their legal representation or next of kin, depending on the study centre, consented on their behalf. Once consent was obtained, patients were enrolled in the study. A copy of the consent form was retained in the study trial site and a copy given to the participants. PRs documented patients’ inclusion in the study in their medical notes.
Data collection
Recruitment was by PRs across the six sites. PRs were made up of physicians, pharmacists and nurses with training and experience in geriatric medicine. Patients’ data were collected at two time points, that is, time of recruitment and time of discharge or day 14. All data collected were entered into an electronic case report form (eCRF). Baseline data recorded at the time of recruitment included demographics, medical history, medication use, laboratory values and electrocardiogram (ECG). Validated scales were applied to assess prescribing appropriateness [Medication Appropriateness Index (MAI)], cognition [mini-mental state examination (MMSE)], functional abilities (Barthel Index) and burden of morbidities [Cumulative Illness Rating Scale-Geriatrics (CIRS-G)]. Nutritional status was assessed using the Mini-Nutritional Assessment (MNA). At baseline, potential prevalent AEs were recorded.
Patients were reviewed again at the time of discharge or day 14. At this time point, new diagnoses and an updated list of medications were recorded. Patients’ medical and nursing notes, along with medication records, were retrospectively reviewed to assess for incident AEs according to the trigger list of potential ADRs, and also to follow up on prevalent AEs recorded at the time of recruitment. If patients were still hospitalized at day 14, they were reviewed again at time of discharge to get an up-to-date list of medications.
Baseline information was used to calculate a predictive ADR score using the recently developed prediction tool, called the Adverse Drug Reaction Risk in Older Persons (ADRROP) scale, as well as the published GerontoNet ADR risk score. 13 The accuracy of these two scoring systems was then compared using the study data.
Screening process
A process was developed to ensure that patients were screened for eligibility in an unbiased fashion. Patients were systematically screened according to a fixed admissions list. Some centres received this list alphabetically or according to hospital ward whilst others received it by time of admission alone. All centres were consistent in their agreed approach from the start to the end of the study.
Adverse drug reaction endpoint adjudication process
A robust system was put in place to ensure all AEs were adjudicated in an unbiased fashion. The above mentioned trigger list ensured that the majority of potential ADRs were reported to the adjudication committee; adjudication of potential endpoints was undertaken by a quorum of the six clinical site principal investigators (PIs), excluding the PI from the site where the potential ADR had occurred. The work of this committee was directed by an endpoint liaison officer (LO), based within the data management centre at the Health Research Board Clinical Research Facility in Cork. The PEPAC adjudicated the potential drug-related causality of the event using standardized criteria. Incident ADRs of nontrivial severity that were judged to be probably or certainly drug related were accepted as a primary study endpoint. See Supplementary information Figure 1 for a review of this process.
Statistical analysis
Categorical variables were described by the counts and percentages of patients in each level. Unimodal, symmetric continuous variables were described by their means and SDs. All other continuous variables were described by their medians and IQR. CIs for the primary outcome and any other binomial variables were constructed using the Clopper–Pearson interval. Tests of dependence between the primary outcome and categorical variables were conducted using Pearson’s chi-squared test with a continuity correction. Tests of dependence between the primary outcome and continuous variables were conducted with Welch’s two-sample t test with unequal variances, or the Mann–Whitney U tests, as appropriate.
The validity of the previously developed ADRROP and GerontoNet tools were examined in this sample using a logistic regression classifier. Results were reported as odds ratios (ORs) and 95% confidence intervals (CIs) for each ADRROP predictor. The overall predictive utility of the model was evaluated using the area under the receiver–operator characteristic curve.
Ethical approval
Ethical approval was sought and obtained by the local research ethics committees across all six sites.
Results
Over 18 months, a total of 644 patients were recruited across the six centres [Cork: 149 patients (23.1%); Reykjavik: 110 patients (17.1%); Aberdeen: 126 patients (19.6%); Madrid: 125 patients (19.4%); Ghent: 71 patients (11%); Ancona: 63 patients (9.8%)]. Patient characteristics are described in Table 2. Patients were evenly split by sex and overwhelmingly of White ethnicity. Almost half were former smokers, and <15% had completed third-level education. Over 80% of cases were medical (as distinct from surgical) admissions. The median number of chronic conditions being treated at enrolment was 5 [interquartile range (IQR) 4–6], with eight or more chronic conditions present in approximately 10% of cases. The mean number of mediations taken by patients was 9.9 [standard deviation (SD) 3.8], and was strongly correlated with the number of chronic conditions (r = 0.54, p < 0.0001).
Patient characteristics (n = 644).
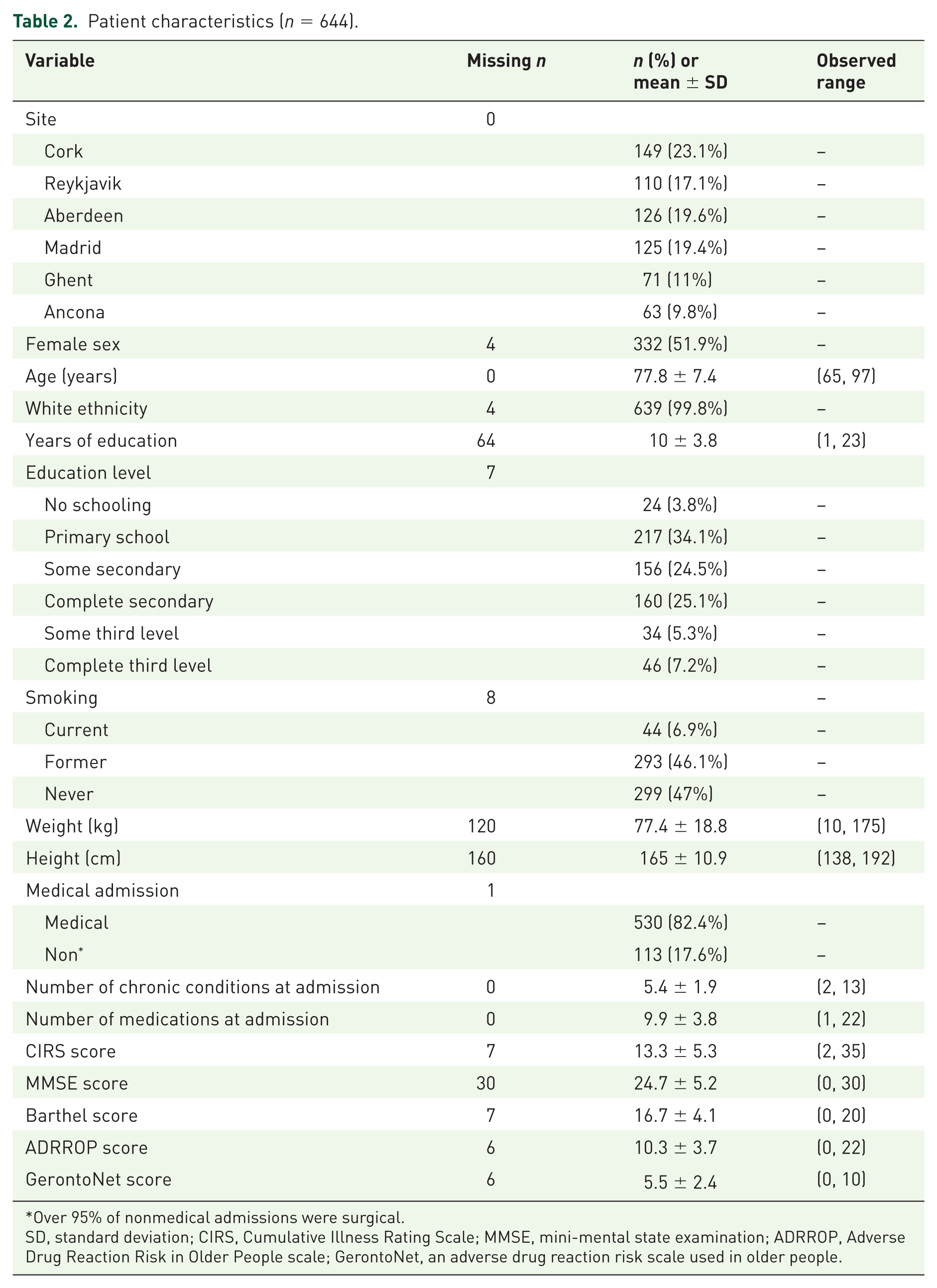
Over 95% of nonmedical admissions were surgical.
SD, standard deviation; CIRS, Cumulative Illness Rating Scale; MMSE, mini-mental state examination; ADRROP, Adverse Drug Reaction Risk in Older People scale; GerontoNet, an adverse drug reaction risk scale used in older people.
There were 732 AEs in total recorded [Cork: 62 (8.5%); Reykjavik: 144 (19.7%); Aberdeen: 196 (26.7%); Madrid: 171 (23.4%); Ghent: 107 (14.6%); and Ancona: 52 (7.1%)]. These events occurred in 382 patients [Cork: 39 (10.2%); Reykjavik: 76 (19.9%); Aberdeen: 91 (23.8%); Madrid: 86(22.5%); Ghent: 56 (14.7%); and Ancona: 34 (8.9%)]. Serum electrolyte disturbance was the most common type of AE (160/732) reported. Only 51/732 AEs (6.96%) were classified as unspecified. The distribution of AE types is given in Table 2. Of all 732 AEs, 363 were incident. As expected, there was considerable heterogeneity across sites (χ2 = 255.65, df = 60, p value < 0.001; Table 3).
Percentage of patients experiencing at least one adverse drug reaction (ADR), with 95% confidence intervals.
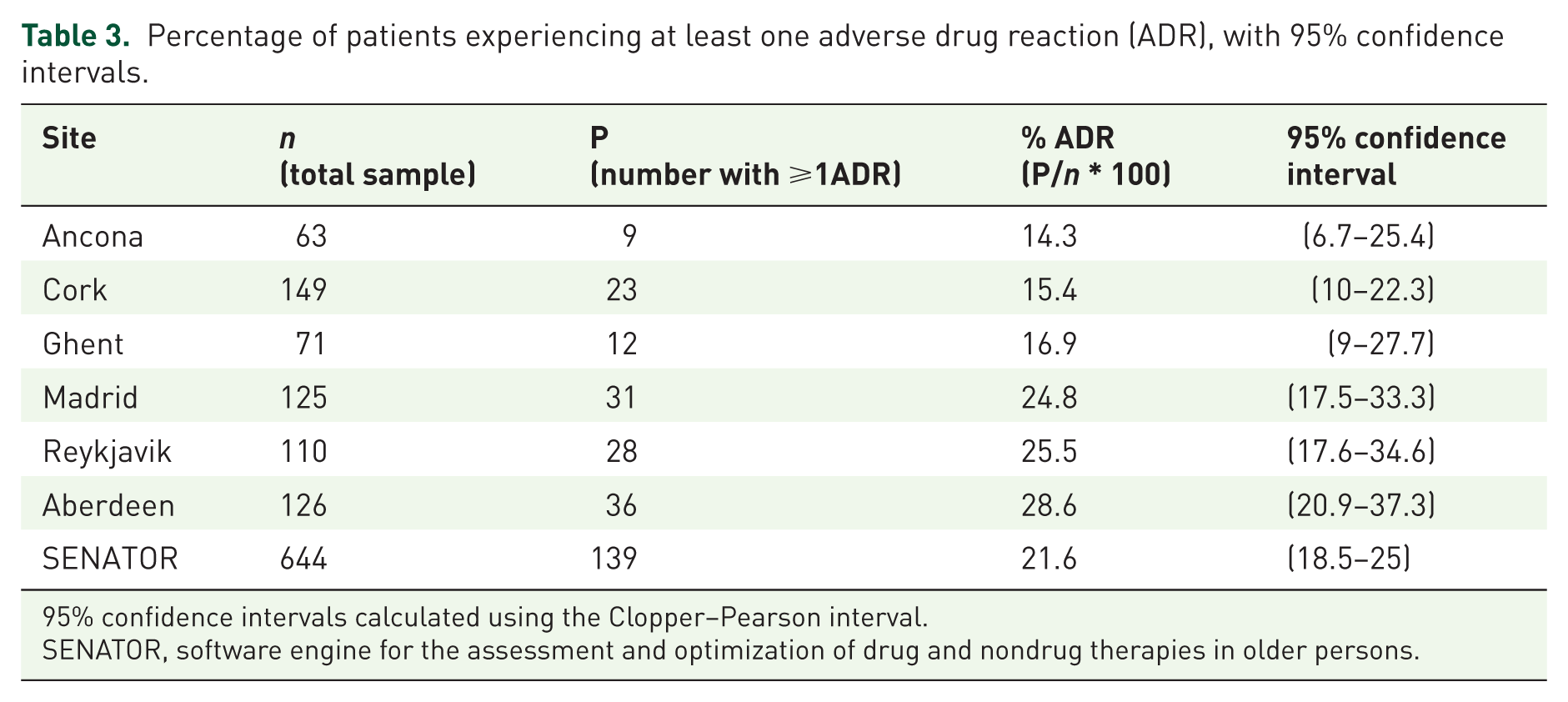
95% confidence intervals calculated using the Clopper–Pearson interval.
SENATOR, software engine for the assessment and optimization of drug and nondrug therapies in older persons.
An AE was deemed to be an ADR when any suggestion of a link to a certain drug was adjudicated as being probable or certain. After adjudication, the great majority of events were classified as having been probably or possibly caused by a drug (Supplementary information Figure 2), with some heterogeneity across sites (χ2 = 88.567, df = 20, p value < 0.001). For this study, the patient-level primary outcome was the presence of one or more ADRs defined in this manner. Based on this definition, 176 of 363 incident AEs (48.5%) were adjudicated to be ADRs. Of these ADRs, approximately 50% occurred within the first 3 days post-study enrolment and 75% within the first 6 days postenrolment. Out of 644 patients, 139 experienced the primary endpoint (21.6%; 95% CI 18.5–25.0%). The clinical site with the lowest percentage of patients experiencing an ADR was Ancona (9/63; 14.3%; 95% CI 6.7–25.4%), while the site with the highest was Aberdeen (36/126; 28.6%; 95% CI 20.9–37.3%). Serum electrolyte abnormality was the most common ADR reported (32/176; 18.2%). The full set of clinical site estimates and 95% CIs are given in Table 3. Across the six sites, there was no significant difference in the relative percentage of medical patients experiencing one or more ADRs (21.5%) versus nonmedical (22.1%) patients (χ2 < 0.001, df = 1, p value = 0.99), although the absolute numbers varied considerably between sites. The proportions of patients experiencing one or more ADR according to clinical site and admission type (i.e. medical versus nonmedical) is presented in Supplementary information Figure 3. There was no statistically significant difference in the occurrence of ADRs between men and women (19.1%; 95% CI 14.9–24.0% versus 24.0%; 95% CI 19.5–29%, χ2 = 2.0125, df = 1, p value = 0.16) (Supplementary information Table 5). The mean age of patients experiencing an ADR (79.2 years) was 1.8 years older than those who did not (77.4 years), p value = 0.013.
The ADRROP and GerontoNet ADR risk scales were applied to this population to assess their ability to predict ADRs in this patient cohort. The area under the curve (AUC) was 0.61 for the ADRROP scale and 0.60 for the GerontoNet ADR risk scale (Supplementary information Figure 4), that is, ADRROP correctly predicted ADR occurrence in 61% of patients and GerontoNet in 60% of patients. Thus the predictive ability of both tools was limited in this patient cohort.
Discussion
This study reports an incident ADR rate of 21.6%. This is higher than in most previous studies. It reports a similar incident ADR rate to that previously reported from Cork. 4 This is the first study to examine simultaneous incident ADR rates across six European centres. We contend on the basis of our previous observational studies4,17 that the use of an AE trigger list enables a clear, structured approach to assessing common ADRs in this older complex multimorbid population. Prior to this study, a shorter 10-AE trigger list had only been used in Cork; 4 the expanded 12-AE trigger list has been employed with the aim of achieving a higher ‘capture rate’ of ADRs.
It became evident early in the study that some of the planned tools chosen as part of data collection were not suitable for use in the proposed trial. The MAI was time consuming and duplicated some of the work of the planned software intervention for the RCT, for example, highlighting drug–drug interactions, drug–disease interactions and duplication of drugs. The time burden of its deployment would substantially prolong baseline data acquisition and delay randomization. Therefore, it was decided to avoid the MAI altogether in the RCT. Similarly, the MNA added participation burden with challenges around recording patients’ current weight, height and potential weight loss in the preceding months, thus it was decided that this tool should no longer be used.
The present study also highlighted many challenges that exist around ADR ascertainment and has allowed us to refine our ADR processes for the future RCT. The first challenge was to ensure that prevalent ADRs were clearly distinguished from incident ADRs. As the feasibility study was purely observational, one could have argued that all events that occurred in hospital, regardless of the time of recruitment into the study, should be included in the study. Conversely, one could insist that only adverse events identified following enrolment into the study should be classified as incident events. Therefore, in the interest of data accuracy and consistency, we retrospectively reviewed all patients’ data at the end of the trial to confirm all potential incident ADRs reported were indeed incident relative to the study, that is, were defined as those that occurred postenrolment. The reason for this was to get an accurate reflection of the number of incident ADRs that could potentially be altered by the intervention in the forthcoming RCT. In the RCT, we have clearly defined prevalent events as those that occur before hospitalization or in hospital prior to randomization, and incident events as those that occur after randomization.
For this study, ADRs were defined as discrete events. This approach introduced a level of subjectivity in distinguishing between repeat events, especially for laboratory-based adverse events such as hyponatremia. It was problematic for researchers to define when an event started and when it finished. Attempting to accurately and reproducibly distinguish between recurrent events was extremely time consuming. This raised concerns for the ability to reliably compare absolute numbers of discrete events across sites and investigators. This led us to examine our events as processes and to report the occurrence of one or more incident events in our results while noting the overall severity of the ADR using the Hartwig and Siegel scale. The subjective interpretation of the endpoint (in this case, incident ADRs) is a major challenge to internal validity in an open-labelled study and for our forthcoming RCT. Therefore, we will view ADRs as processes, record the consequences that arise as a result of the event(s) and we will not attempt to define exactly the number of discrete occurrences of the given putative ADR. For example, a person who falls secondary to a medication but has no trauma is different from the person who falls, suffers a fractured hip, has an increased length of stay and subsequently transitions to nursing home care. We will capture linked events in patients through the trigger list as well as changes in patients’ medications during admission and their length of stay. All patients will participate in a 12-week follow-up questionnaire which will record their engagement with healthcare professionals posthospital discharge, as well as their medications and current living arrangements.
Although we were unable to accurately report every single occurrence of each type of ADR, we did note that on many occasions, separate types of ADRs were potentially causally related and not independent of each other, for example, a patient presenting with diarrhoea secondary to a medication which subsequently led sequentially to an acute kidney injury, orthostatic hypotension and a fall. Our data collection processes were not set up to capture this sequence of events in its entirety, that is, we were not able to delineate between events that were independent of others and those that were dependent. When reporting the frequency of ADRs, independent events should be reported separately to those that are dependent. This has now been addressed for the SENATOR RCT so that we will be able to report the percentage of independent ADRs, type of ADRs, as well as the number, potential relationships between events and overall burden of ADRs. This has not been well addressed in the literature to date.
This study found that many ADRs occurred early in the patients’ admissions. Our extended time window of 72 h allowed for greater recruitment, and our definition of incident ADRs meant that these events within 72 h were not classified as incident events. Thus, it is likely the true incidence of hospital-acquired ADRs is in fact higher than we are reporting here and that to avoid missing these events in future studies, patients should be recruited as early as possible into their admission, ideally within 24 h of admission. Consequently, we have refined our screening process for the upcoming RCT, that is, patients will be recruited within 60 h of admission.
Finally, the variability between the ADR rates between medical and surgical specialities, within medical and surgical specialities and across all six sites, along with the modest sample size and the failure of the ADRROP predictive tool to accurately predict ADRs, means that for the SENATOR RCT, potential differences in baseline ADR rates cannot be controlled. Whilst it is not entirely clear why substantial variability was observed across the six clinical sites and within the core categories of medical and surgical admissions, it likely relates to the differences between the specialty pool in which recruitment took place; for example, in Cork, no orthopaedic patients were enrolled, a population where the ADR rate is relatively high. 6 Furthermore, the degree of specialist geriatric input postenrolment was likely variable. This relates to different clinical customs and practices and pathways of care across the clinical sites. To account for this in the RCT, geriatric input postrecruitment will be recorded. In addition, the ADR predictive power of ADRROP and GerontoNet ADR risk scales were limited in this patient population (Figure 4, Supplementary material). For the RCT, this means those patients who experience ADRs cannot be accurately identified in advance using either ADRROP or GerontoNet ADR risk scales. The variability in ADR rates and the failure of these risk scales to accurately predict ADRs, means that for the RCT, randomization of patients will occur at an individual patient level rather than by cluster, defined by ADR risk.
The main strength of this study was the robust ADR adjudication process. This process, in conjunction with the AE trigger list meant that all nontrivial probable or certain incident ADRs were reported in a reliable and unbiased fashion. Throughout this study, to complement this process, regular monthly teleconferences were held for all site researchers to discuss any AE that generated uncertainty. This continuous learning allowed the primary researchers to address any challenges that arose in real time. This process will be retained for the SENATOR RCT. This study also allowed us to address any potential challenges around ADR assessments and to ensure that our methodological processes are robust and fit for purpose in the SENATOR RCT.
This feasibility study accomplished its main aims which were to establish the incident ADR rates across the six trial centres, which will not only guide power calculation in the SENATOR RCT but also guide the randomization process. The upcoming trial will examine the efficacy of the SENATOR software as an intervention for preventing ADRs in older multimorbid people who are hospitalized with an acute illnesses. The SENATOR software, for patients in the intervention arm, will advise on potential drug–drug interactions, potential drug–disease interactions and recommend on how to prevent incident delirium through nonpharmacological measures. Finally, the software will select and display the relevant STOPP and START criteria relevant to each intervention patient. The working hypothesis for this trial is that the SENATOR software intervention will significantly reduce incident ADRs compared with standard pharmaceutical care of these patients during an acute hospitalization. The SENATOR trial will involve randomization of 1800 older multimorbid patients and is expected to conclude and report in 2018.
Footnotes
Acknowledgements
This work was supported by European Commission’s Seventh Framework Programme (FP7/2007-2013) (grant number 305930) as part of the SENATOR project. Health Research Board Clinical Research Facilities at University College Cork (HRB CRF-C) The patients and staff of the participating hospitals:
University College Cork, National University of Ireland Cork (UCC) Universiteit Gent (UGENT) Servicio Madrileno de Salud (SERMAS) Istituto Nazionale di Ripose E Cura per Anziani (INCRA) Landspitali University Hospital (LUH) Grampian Health Board (NHS Grampian) The staff of the following organizations:
ClinInfo S.A. (CLIN) ARRTIC International Management Services University of East Anglia (UEA)
Funding
This work was supported by European Commission’s Seventh Framework Programme (FP7/2007-2013) (grant number 305930) as part of the SENATOR project.
D. O’Mahony is a coprivate investigator in the OPERAM project funded by the EC under Horizon 2020. OPERAM will examine the efficacy of STRIP-Assistant software in the prevention of medication-related admissions to hospital and associated excess healthcare costs.
Conflict of interest statement
The authors declare that there is no conflict of interest.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.