
Abstract
In response to good-intentioned efforts to correct the traditional undertreatment of pain, opioid prescribing experienced a dramatic increase over the past decade. But there is now concern that the pendulum has swung too far in the opposite direction, with a rise in morbidity and mortality associated with prescription opioid misuse and abuse. Yet despite potential problems, opioids are a mainstay for the treatment of pain and are an important component of a comprehensive pain management strategy. Therefore, the overall goal of pain management is to decrease pain and to improve patient functioning and quality of life, while monitoring for adverse effects or aberrant behaviors. That is, to balance effective chronic pain management with appropriate use of opioids. Toward this end, several abuse deterrent strategies (formulations) have been, and continue to be, developed. Several major ones are reviewed here. These products should be a first step in trying to address diversion and abuse in a manner that does not discriminate against any particular patient and aligns with universal prescribing precautions. They should also comprise only one aspect of an overall opioid risk management plan.
Introduction
Overview of chronic pain
Most recent estimates suggest that 100 million Americans suffer from chronic pain each day [Institute of Medicine, 2011]. To put this into perspective, patients with chronic pain outnumber those with cancer, diabetes and heart disease combined [The American Academy of Pain Medicine, 2013]. Many patients receive inadequate or ineffective pain management. As a result, they often experience a myriad of psychological and physical debilitations, resulting in a reduction in overall quality of life [Stewart et al. 2003; Gaskin and Richard, 2012]. Undertreated pain also can be a burden on society, draining healthcare resources and driving up healthcare costs [Gaskin and Richard, 2012]. In an effort to fight pain, various stakeholders championed increased access and options for pain medications, including opioids.
Overview of opioid use/abuse
Opioids have increasingly become a mainstay treatment option for pain, recommended by various pain societies and stakeholders. This has led to a large increase in consumption over the past two decades. Prescription fills from 1994 to 2003 increased from 222 to 354 million, while sales of opioids from 1997 to 2007 increased between 280% and 1293%, depending on the opioid [Manchikanti et al. 2010]. This increase in consumption has also led to an increase in the nonmedical use of prescription pain relievers. Results from the 2012 Substance Abuse and Mental Health Services Administration’s National Health Survey reported that 4.9 million Americans over the age of 12 were nonmedical users of prescription pain relievers, slightly higher than the 4.5 million in 2011 [SAMHSA, 2012]. In addition, deaths due to drug overdoses have increased substantially since the early 1970s, with approximately 27,000 deaths reported in 2007 and approximately half due to opioid analgesics [Centers for Disease Control, 2012].
Opioid risk management
Objectives
Opioids play an important role in the treatment of chronic pain [Trescot et al. 2008]. Despite barriers, there is a need to balance effective chronic pain management with appropriate use of opioids, which in its simplest sense refers to the effort to maintain appropriate access to opioid therapy while minimizing harms [Katz et al. 2007]. The goal of pain treatment is to decrease pain and improve functioning while monitoring for adverse effects or aberrant behaviors [Gourlay et al. 2005]. Prevention of opioid abuse and misuse has been difficult over the past few decades. It is often difficult for physicians to try and strike a balance between effective pain treatment and risk management. In order to adequately manage chronic pain, risk stratification prior to initiating therapy should be considered.
Pain management decision tree
Physicians should individualize treatment in every case, using nonopioid analgesics, opioids on an as-needed basis and/or combination products, and chronic opioid therapy in a progressive plan of pain management. Prior to any pain regimen, a thorough pain assessment should be conducted. A comprehensive pain assessment should evaluate: onset and duration of pain; location, description and intensity of pain; any aggravating or relieving factors; previous treatment; and the impact of the pain. After completing a comprehensive assessment, it is important to screen specifically for risk and safety considerations to determine if the patient has any pre-existing conditions or risk factors, including abuse, misuse and addiction. Exploration of various treatment options should be considered and discussed with the patient.
Abuse-deterrent strategies
In general, patients with moderate to moderately severe chronic pain require pain relief throughout their entire day. Extended-release formulations of opioids that are indicated for moderate to severe, around-the-clock pain are viable options. However, many of the early versions of these formulations were easily abusable, especially through various tampering techniques. Not all extended release opioids have abuse- or tamper-resistant properties and thus opioid products that may reduce abuse should be considered first-line treatment. Most preferable would be those with multiple deterrent strategies such as pharmacologic, aversive component, physical or deterrent packaging (Figure 1). Some products might utilize transdermal pain patches.
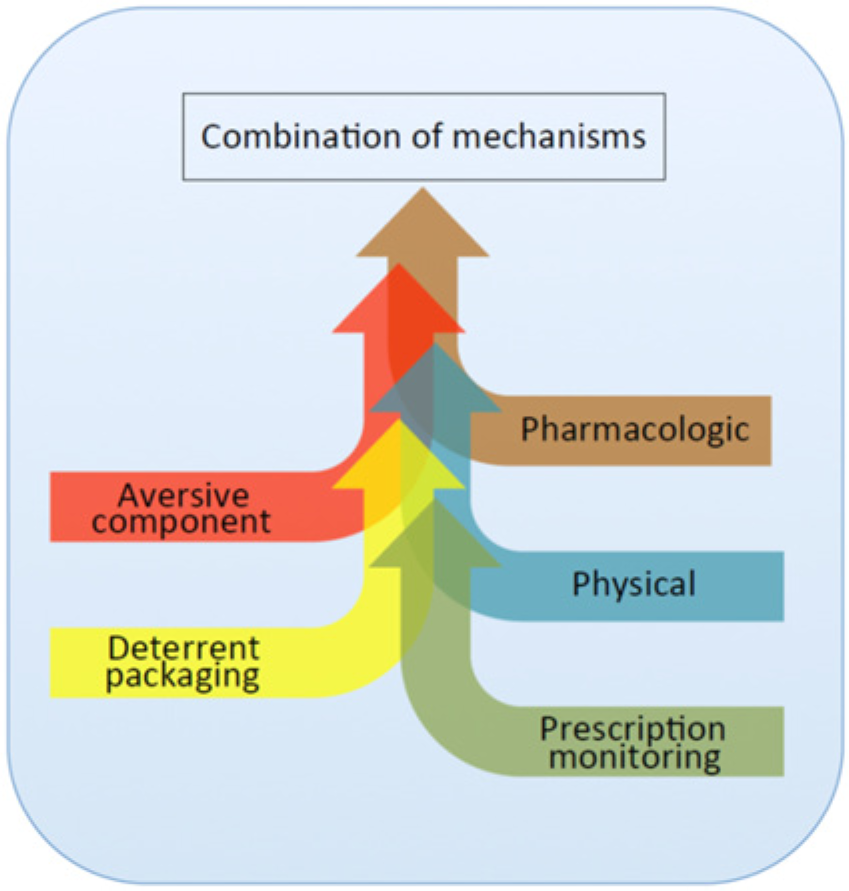
Abuse-deterrent strategies.
Extended-release options
Combination products containing opioid agonists and antagonists are not a new concept, dating back to the 1970s when naloxone was incorporated into pentazocine to discourage use by recreational drug abusers. Today, several newer products are utilizing the opioid antagonist naltrexone. Naltrexone can be sequestered at the core of the formulation such that it is only released upon tampering. When such formulations are taken properly, the patients experience pain relief and no opioid withdrawal effects. However, upon tampering (e.g. crushing) naltrexone is released along with the opioid agonist and blocks the opioid agonist effects. Three such products have been described in the literature, two have undergone clinical testing, and one has been approved by the United States Food and Drug Administration (FDA). A summary of their clinical development is discussed below.
Morphine + naltrexone
The formulation of opioid agonist and antagonist approved by the FDA is EMBEDA®, which is an extended release morphine with sequestered naltrexone. Naltrexone is sequestered within the core of the pellets in the formulation and is designed to be released upon tampering (chewing or dissolving). Sufficient naltrexone can be released to precipitate withdrawal in opioid-dependent individuals, raising concerns among physicians and regulators [Ruan et al. 2010; Setnik et al. 2013; US FDA, 2013a]. Combining morphine and naltrexone has been reported to provide analgesic efficacy with a good safety profile and reduction in drug likability [Pfizer Inc., 2009; Stauffer et al. 2009; Katz et al. 2010a, 2010b; Webster et al. 2010, 2011; Johnson et al. 2012]. At the time of writing this article, the marketed morphine + naltrexone product has been withdrawn from the market due to stability problems. Efforts are underway to relaunch the product in 2014.
Efficacy
In a phase II, multidose bioequivalence, efficacy and safety study of morphine + naltrexone versus extended release morphine for chronic osteoarthritis of the hip or knee, various efficacy parameters were evaluated in 113 enrolled patients [Katz et al. 2010b]. They included pain scores, Western Ontario and McMaster Universities (WOMAC) Osteoarthritis Index and Patient Global Assessment of Medication. Morphine + naltrexone provided similar pain relief and overall patient satisfaction similar to extended release morphine. Both products had similar Brief Pain Intensity scores at the end of the 14 day treatment, with scores of 2.3 versus 2.4 (p = 0.31) for morphine + naltrexone versus extended release morphine, respectively. Changes in WOMAC scores for pain, physical function and composite scores from baseline to end of treatment were also similar between both treatment groups. More patients on morphine + naltrexone reported greater satisfaction with medication than those on extended release morphine (91.5% versus 78.9%). The authors concluded that incorporation of naltrexone did not interfere with morphine’s efficacy and that morphine + naltrexone provides similar pain relief as extended release morphine.
In a pivotal phase III double-blind, randomized, 12-week study utilizing patients with chronic osteoarthritis, efficacy between morphine + naltrexone and placebo was tested [Katz et al. 2010a]. Primary efficacy was mean change in the weekly diary Brief Pain Inventory (BPI) to end of study. Secondary endpoints included additional BPI scores as well as WOMAC for Osteoarthritis. The mean change from baseline to end of study pain score between morphine + naltrexone versus placebo was −0.2 ± 1.9 versus 0.3 ± 2.1 (p =0.045). The change in baseline to other points during the maintenance phase were also significantly in favor of morphine + naltrexone (p < 0.05). In addition, WOMAC composite score was superior to placebo at most visits throughout the study. The authors concluded that morphine + naltrexone provided effective pain relief and incorporation of naltrexone did not compromise morphine’s efficacy.
Safety
Long-term safety of morphine + naltrexone was evaluated in a 52-week phase III study [Webster et al. 2010]. A total of 465 subjects received at least one dose of study drug, with 65.7% prematurely discontinuing from the study due to adverse event (23.7%), noncompliance (13.7%) or lack of efficacy (8.4%). Safety assessments included adverse events (AEs), laboratory assessments and the Clinical Opiate Withdrawal Scale (COWS). Investigators recorded AEs at each study visit and recorded intensity, relationship to study drug, action taken and outcome. Distribution of AEs was similar to that of randomized controlled studies and was consistent with most common opioid related AEs. In addition, at the end of the safety study, plasma naltrexone levels were too low to be measured in 89% of blood samples assayed; exposure to naltrexone or its metabolite was not clinically meaningful after 12 months. Throughout the clinical program, 1251 subjects were exposed to one dose of morphine + naltrexone. Serious adverse reactions included respiratory depression, respiratory arrest, apnea, circulatory depression, cardiac arrest, hypotension and shock [Pfizer Inc., 2009]. Common AEs were drowsiness, dizziness, constipation and nausea. Common AEs in a phase III pivotal trial leading to discontinuation included nausea, constipation and vomiting.
Morphine + naltrexone pharmacokinetics and safety was also tested in the presence of alcohol in a phase I, randomized, open-label, four-way crossover study in healthy adult volunteers (n = 32) [Johnson et al. 2012]. Mean time to maximum concentration (C max) values for morphine + naltrexone with co-ingestion of either 4% or 20% alcohol were similar to mean C max values of subjects administered morphine + naltrexone and water; however, 40% alcohol had a mean C max value approximately twofold greater than morphine + naltrexone administered with water. Morphine + naltrexone with 40% alcohol had a clinically significant faster rate of exposure as demonstrated by an earlier release of morphine with a median t max of 4 hours. Co-ingestion of alcohol may result in increase in plasma levels and potentially fatal overdose of morphine. A total of 23 (72%) volunteers experienced an AE – the most common included headache, nausea, vomiting and dizziness. The methodology for identifying these AEs was not reported.
Drug liking
The pharmacodynamic (PD) effect of naltrexone in the setting of crushed morphine + naltrexone has been examined in two clinical studies: a phase I oral PD study and a phase I simulated intravenous (IV) PD study. The phase I oral PD study was a randomized, double-blind, triple-dummy, four-way crossover where 32 nondependent recreational opioid users received 120 mg morphine + naltrexone whole or crushed, 120 mg immediate release (IR) morphine and placebo [Stauffer et al. 2009]. A significant reduction in the maximal effect for drug liking was observed for all treatment combination comparisons (p < 0.001) except for morphine + naltrexone whole versus morphine + naltrexone crushed (adjustedp = 0.875). The area under the curve to 24 hours for drug liking was statistically significantly different for treatment comparisons of morphine versus placebo, morphine + naltrexone whole, and morphine + naltrexone crushed (p < 0.015). Overall 87.5% of subjects had some degree of reduced drug liking after receiving crushed morphine + naltrexone, while 12.5% had no reduction in drug liking. Some degree of decreased euphoria with crushed morphine + naltrexone compared with IR morphine was shown by 69% of subjects.
The phase I simulated IV PD study was a randomized, double-blind, placebo-controlled 3-way crossover trial in 28 nondependent recreational opioid users [Webster et al. 2011]. Subjects were administered 30 mg IV morphine alone or in combination with 1.2 mg of IV naltrexone to simulate parenteral use of crushed morphine + naltrexone combination product. The primary objective was to determine the effect of naltrexone on the subjective effects of morphine when given intravenously. In addition, secondary assessments included the Drug Effects Questionnaire, Cole/ARCI Stimulation Euphoria Scale and plasma concentrations of naltrexone. The combination of morphine + naltrexone resulted in 71% of subjects reporting some degree of reduced euphoria compared with morphine alone, while 29% had no reduction in euphoria. Based on Question #5 of the Drug Effects Questionnaire, subjects taking morphine alone were nearly three times more likely to answer ‘feeling high’ than those on morphine + naltrexone or placebo (29.8 versus 85.2 versus 0.00 mm). Secondary endpoints including the Cole/ARCI Stimulation Euphoria scale (13.7 versus 27.8 mm versus 1.3) and drug-liking scores (38.9 versus 81.4 versus 0.0 mm) all were significantly (p < 0.0001) diminished in subjects receiving morphine + naltrexone versus morphine alone or placebo.
To assess morphine + naltrexone’s real abuse deterrent potential as well as additional safety concerns surrounding opioid withdrawal with this formulation [Romach et al. 2013], postmarketing epidemiological studies are currently needed. Until the product returns to market, its ability to deter abuse and prevent abusers from manipulating it remains to be evaluated.
Oxycodone + naltrexone
Oxycodone plus ultra-low-dose naltrexone has been tested at a variety of doses ranging from 2.5 to 40 mg for oxycodone with fixed doses of naltrexone of 0.001 or 0.0001 mg naltrexone. The combination product was previously branded under OXYTREX™. The product to date has not been approved by the FDA due in part to a failed phase III trial. Currently, there are a number of ongoing clinical trials listed on the National Institutes of Health clinical trial website (ClinicalTrials.gov) that are testing oxycodone + naltrexone combination (ALO-02).
Efficacy
Two published trials have examined the efficacy of this product in osteoarthritis and low back pain [Chindalore et al. 2005; Webster et al. 2006]. A second phase III trial was not published due to cessation of clinical development based on the results, but was presented at one of the annual pain conferences supported by the American Pain Society. In a phase II, 3-week, double-blind placebo- and active-control escalation study, 362 patients with osteoarthritis were randomized to one of 4 treatment arms which included oxycodone twice daily (bid), oxycodone + naltrexone four times a day (qid), oxycodone + naltrexone bid, and placebo [Chindalore et al. 2005]. Primary endpoint was the change in baseline of pain intensity with secondary endpoints including global assessment of pain, quality of analgesia, pain control and WOMAC Index. At the end of 3 weeks, patients taking oxycodone + naltrexone bid experienced a 39% reduction in pain intensity compared with a 21.5% (p < 0.001), 24.6% (p = 0.006) and 26% (p = 0.003) reduction in patients taking placebo, oxycodone qid and oxycodone + naltrexone qid, respectively. Oxycodone + naltrexone bid was also superior to placebo in other secondary assessments. Quality of analgesia for all active treatments was significantly improved at week 3 compared with placebo (p = 0.002). Duration of pain control each day was also significantly better than placebo during week 3 (p = 0.05). In addition, the patient’s global assessments (p = 0.04) and the WOMAC Osteoarthritis Index total score (p = 0.03) were significantly improved for all active treatments versus placebo.
There were 719 patients with low back pain who were randomized to one of 4 treatment arms in a phase III, double-blind, placebo- and active-control dose escalation trial. Treatment groups consisted of placebo, oxycodone bid, oxycodone + naltrexone qid, or oxycodone + naltrexone bid [Webster et al. 2006]. Patients were initiated on a dose of 10 mg/day of oxycodone with or without naltrexone and escalated to a maximum of 80 mg/day. Optimal doses were maintained for 12 weeks. At the end of 12 weeks, all active treatment groups significantly reduced pain intensity compared with placebo (p < 0.05), improved the physical component score of the SF-12 (health survey), quality of analgesia, and the global assessment of study medication. However, there were no significant differences in any of these assessments across the active treatment groups. Even though there was no difference in regards to reduction in pain intensity, subjects receiving oxycodone + naltrexone had an overall lower drug use of oxycodone compared with oxycodone alone (p = 0.03). Oxycodone + naltrexone bid. patients reported 55% less physical dependence than patients on oxycodone as reported by the Subjective Opiate Withdrawal Scale (SOWS) after 24 hours.
There were 775 patients with osteoarthritis enrolled into a second phase III, double-blind, placebo- and active-controlled trial to assess oxycodone + naltrexone in reducing physical dependence [Spierings et al. 2006]. Patients received a fixed daily dose of oxycodone + naltrexone (20 or 40 mg), oxycodone (40 mg) or placebo for 12 weeks. Endpoints included were physical dependence (primary), as measured by subjective opioid withdrawal scale (SOWS). Secondary endpoints included change in baseline of pain intensity, SF-12, WOMAC, and patient global assessment of medication and pain control. For the primary endpoint, a 22% reduction in physical dependence was found with oxycodone + naltrexone 20 mg; however, statistical significance was not achieved due to the high percentage of dropouts (51%), attributed to inadequate analgesia and adverse effects. There was no significant difference between active and placebo groups in change in pain intensity or change in SF-12, WOMAC, quality of analgesia, pain control or global assessment at the end of treatment.
Safety
The safety profile of oxycodone + naltrexone varied between the three reported clinical studies. In the phase II trial of 362 osteoarthritis patients, AEs which were spontaneously reported by patients or observed by physicians at each visit, between oxycodone + naltrexone and oxycodone included nausea (24% to 39%), constipation (19% to 22%) and dizziness (26% to 32%) [Chindalore et al. 2005]. No significant difference between oxycodone + naltrexone and oxycodone were noted. In the phase III trial of 719 chronic low back pain patients, experience of AEs which were spontaneously reported by patients or observed by physicians at each visit, such as constipation, somnolence and pruritus were significantly (p = 0.01, p = 0.03, p < 0.001, respectively) less in oxycodone + naltrexone bid. compared with oxycodone [Webster et al. 2006]. In the phase III trial of 775 patients with chronic low back pain, 392 patients did not complete the study [Spierings et al. 2006]. Patients in the active treatment groups discontinued primarily due to AEs associated with opioid-related side effects. In addition, inadequate pain relief was also a common reason for discontinuation in the placebo (23.7%) and naltrexone (24.4%) groups. Common AEs consisted of those typical of opioids including dizziness, constipation, dry mouth, nausea, vomiting, somnolence and pruritis. There were no significant differences in incidences of these AEs among active treatment groups.
Abuse potential
Abuse potential of oxycodone + naltrexone was addressed in a double-blind, crossover, placebo-controlled study utilizing 14 experienced opioid abusers. Subjects experienced 7 drug conditions given at least 5 days apart and included placebo, oxycodone 20 mg, oxycodone 40 mg and each oxycodone dose + naltrexone 0.0001 or 0.001 mg naltrexone. Endpoints included visual analog scales for subjective effects, adjective questionnaires, drug versus money questionnaires, ability to identify drug and physiological effects. All oxycodone arms showed significant differences for peak visual analog ratings of ‘liking’, ‘drug effect’, ‘good effects’ and ‘high’. In addition, all oxycodone arms showed significant differences compared with placebo for the adjective questionnaires and drug versus money value rating. However, no significant differences were seen between active treatment groups with or without naltrexone. The authors concluded that the addition of naltrexone did not alter the abuse liability of oxycodone.
ELI216
ELI216 (Oxynal) is a novel oxycodone + naltrexone combination currently in development by Elite Pharmaceuticals (Northvale, NJ). Unlike other products where naltrexone is sequestered with the opioid agonist, ELI216 consists of individual oxycodone beads and naltrexone beads placed inside a gel capsule. The oxycodone is meant to provide effective long-acting pain relief, while upon crushing, chewing, dissolving or extracting, the naltrexone mixes with the oxycodone. The goal is to prevent euphoria, thus deterring abuse. The product initially showed promising results in a phase II study where primary endpoint was blocking of euphoria [Elite Pharmaceuticals, 2007]. However, no published studies are currently available.
Clinical perspective
Chronic pain management is a balance between risk management and effective pain treatment. Identifying patients who may become problematic during pain treatment can be difficult and often cannot be predicted. To minimize such risk, recommendations have been created to help physicians assess patient risk prior to and during chronic pain treatment. These ‘universal precautions’ are a set of 10 steps aimed at improving patient care along with mitigating/containing risk of abuse [Gourlay et al. 2005]. They include pain diagnosis, psychological assessment, informed consent, treatment agreement, pre- and post-intervention assessment of pain, trial of appropriate opioid therapy and adjunctive medication, pain score/level of function reassessment, regular assessment of analgesia, activity, AEs and aberrant behavior, review of pain diagnosis, and documentation. Current abuse deterrent formulations with sequestered opioid antagonists are generally viewed as a highly attractive choice for the various abuse deterrent strategies (Figure 1). Though the true abuse deterrent potential of these opioid agonist + antagonist formulations are yet to be determined, these formulations may align well with the universal precautions, fit within a comprehensive opioid risk management plan and be an appropriate opioid trial choice. In addition, although these abuse deterrent formulations may have higher acquisition costs, the potential societal benefits may justify these costs. [Skinner, 2012]. In order to achieve these benefits, abuse risk mitigation must be implemented across multiple levels to have the best potential impact and use of extended release formulations are an important component of this abuse risk mitigation (Figure 2).
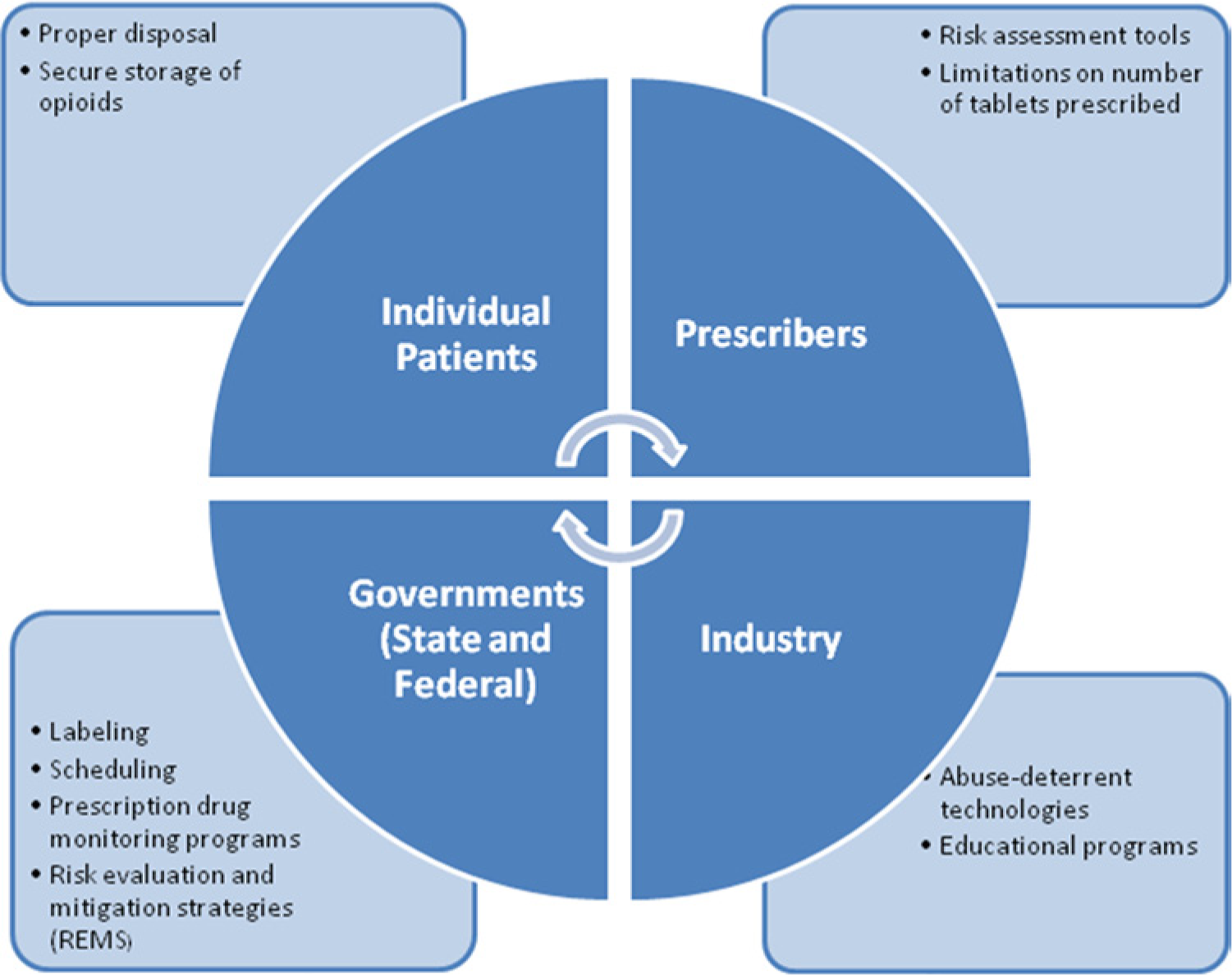
Multiple levels of abuse risk mitigation.
Limitations of reviewed studies
Clinical trials are conducted under widely varying conditions; AEs observed in one trial cannot be directly compared with rates in another trial and may not reflect the rates observed in practice. In addition, AEs may have been reported differently in each trial as well as being suboptimally evaluated, which may be due to how investigators, clinical staff and/or patients observed and reported the events. When using reported efficacy information to make clinical decisions, it is important to fully understand the methodology of each trial. Statistical significance in trials may not necessarily correlate to clinical significance. For example, when comparing morphine + naltrexone with placebo, the number needed to treat to achieve a 30% reduction in pain was 6.8 [Katz et al. 2010a]. Though this was significant in the study, clinically this is only a modest improvement. Similarly, in a superiority study comparing morphine + naltrexone with placebo, morphine + naltrexone treated patients experienced a statistically significant change of 1.6 in WOMAC, though this may not be viewed as clinically significant [Katz et al. 2010b]. Many of the studies did not report results in numbers needed to treat or numbers needed to harm, information that can provide a clearer clinical importance. In addition, these studies did not evaluate the true abuse deterrent potential of these combination products. The FDA has issued a guidance detailing pre- and post-marketing studies as well as four tiers of abuse deterrent product claims [US FDA, 2013b]. Morphine + naltrexone was approved prior to this guidance and therefore its indication does not contain any abuse deterrent language. Although there is evidence to suggest that newer formulations of sustained release oxycodone reduces abuse [Butler et al. 2013; Severtson et al. 2013], solid conclusions regarding the ability of oxycodone + naltrexone and even morphine + naltrexone to reduce and deter abuse cannot be made without these critical pre- and post-marketing studies.
Conclusion
In response to a dramatic rise in prescription opioid misuse and abuse, formulations have been developed that are designed to deter or mitigate such inappropriate behavior, while at the same time allowing legitimate use by chronic pain patients to continue without stigmatization. Several strategies have been utilized, one of which is the combination of an opioid receptor antagonist within the formulation. Although not a new concept per se, several of the newer formulations are incorporating the opioid antagonist naltrexone. Naltrexone can be sequestered at the core of a formulation, so that it is only released upon tampering. It is not released during normal therapeutic use and therefore does not inhibit analgesic effectiveness. When such formulations are taken properly, patients experience the usual pain relief and no opioid withdrawal due to the presence of the unreleased naltrexone. However, if tampering is attempted (e.g. by crushing or solvation), naltrexone is released together with the opioid agonist and blocks the opioid agonist effects. While by no means abuse-proof, these products can be an abuse deterrent or a mitigant to those seeking to misuse or abuse opioids by tampering with legitimate opioid prescriptions. Physicians might consider incorporating these formulations into their overall opioid risk management approach after careful consideration and appropriate weighing of the benefits and risks for each individual patient.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
R.T. is an employee of NEMA Research, a contract research organization that is contracted by various pharmaceutical and biotechnology companies in the area of pain management. NEMA Research was not paid to prepare this manuscript. R.B.R. is a former employee of Johnson & Johnson and former or present speaker, consultant and/or basic science investigator for (abbreviated names) Adolor, Alteon, Ampio, Asta Medica, Discovery Research Consultants, Endo, Galleon, Grünenthal, Inspirion, Johnson & Johnson, Kirax, LaboPharm, LAPID, Mallinckrodt/Covidien, Novartis, Onconova Pain Therapeutics, Pfizer and Purdue Pharma, but receives no royalty (cash or otherwise) from the sale of any product. J.V.P. is a consultant, research and/or speaker for ENDO, Grünenthal, Janssen, Kirax, Luitpold, Mallinckrodt, Merck, Mundi Pharma and Purdue Pharma. He received no funding for his participation in this manuscript and receives no royalty (cash or otherwise) from the sale of any product.