
Abstract
Used mainly for the management of neuropathic pain, pregabalin is a gabapentinoid or anticonvulsant that was initially developed as an antiepileptic agent. After more than a decade of experience with pregabalin, experience and studies have shown that the adverse effect profile of pregabalin is well tolerated for the management of neuropathic pain and other conditions. Its use is associated with benign central nervous system and systemic adverse effects, and there are very limited metabolic, idiosyncratic or known teratogenic adverse effects. Along with its efficacy in particular neuropathic pain conditions, pregabalin’s safety led it to be one of the first pharmacotherapies considered for the management of neuropathic pain. This review discusses the use of pregabalin as well as its potential adverse effects, including the most commonly noted features of sedation, dizziness, peripheral edema and dry mouth. Although other adverse effects may occur, these appear to be uncommon. The review also discusses the clinical implications of pregabalin’s use for the clinician.
Background
Pregabalin (PGB) is a newer generation gabapentinoid which followed the use of gabapentin (GBP). Originally synthesized over four decades ago [Satzinger et al. 1976], GBP was initially developed for use as an adjuvant antiepileptic drug (AED). However, after its release nearly two decades ago, off-label prescriptions for conditions other than epilepsy make up about 90% of GBP’s use [Tansey, 2004]. This was secondary to limited efficacy in epilepsy as an adjuvant AED, but also because of a series of case reports describing the benefits of GBP in the treatment of neuropathic pain (NeP) [Mellick and Mellick, 1995, 1997; Mellick et al. 1995]. After publication of randomized controlled trials in NeP conditions, GBP became a widely used pharmacotherapy for NeP, despite being off label [Backonja, 1999; Rice and Maton, 2001b].
PGB (Table 1) is a newer gabapentinoid, or AED, with great structural similarity to GBP. Just as with GBP, the use of PGB in epilepsy is limited. Instead, nearly all of PGB’s use is for treatment of NeP [Oteri et al. 2010], for which PGB was more directly targeted than with GBP. In addition, PGB is used frequently in the treatment of anxiety [Feltner et al. 2003; Pande et al. 2003; Pohl et al. 2005]. Although the mechanism of action has not been completely revealed, one known mechanism of action likely contributes to PGB’s efficacy [Bauer et al. 2009], even though other potential mechanisms may also occur [Eroglu et al. 2009].
Pregabalin pharmacological summary.

PGB was approved for NeP management in 2004 within the USA and Europe, and PGB has received further indications for various NeP conditions. Of the many treatments available for NeP management [Dworkin et al. 2010; Moulin et al. 2007], gabapentinoids including GBP and PGB are considered as first-line treatment for most clinical guidelines [O’Connor and Dworkin, 2009]. Currently, PGB is indicated for the management of NeP associated with diabetic peripheral neuropathy (DPN) [Arezzo et al. 2008; Bansal et al. 2009; Lesser et al. 2004; Richter et al. 2005; Rosenstock et al. 2004b; Satoh et al. 2011; Tolle et al. 2008], postherpetic neuralgia (PHN) [Achar et al. 2010; Barbarisi et al. 2010; Baron et al. 2009a; Dworkin et al. 2003; Rehm et al. 2010; Sabatowski et al. 2004; Stacey et al. 2008b; van Seventer et al. 2006] and the management of fibromyalgia [Arnold et al. 2008; Crofford et al. 2008; Mease et al. 2008; Ohta et al. 2012; Pauer et al. 2011, 2012] in North America. In the USA as well as in Europe, PGB is also indicated as adjunctive therapy for adult patients with partial onset seizures. PGB is the only medication in Europe approved for the treatment of central NeP. In Europe, it is also indicated for the treatment of peripheral NeP and generalized anxiety disorder, but not for fibromyalgia treatment.
Defined as pain arising from a lesion or disease affecting the somatosensory pathways [Treede et al. 2008] within the peripheral or central nervous system, NeP is a common disorder, impacting on between 4% and 16% of the population [Bouhassira et al. 2008; Torrance et al. 2006; Toth et al. 2009]. Fortunately, PGB is one of several pharmacotherapies used in NeP management which can modulate pain relief and also assist with management of comorbidities.
Mechanism of action, metabolism and pharmacokinetics
The mechanism of action for PGB is not completely understood. As the S-enantiomer of 3-(aminomethyl)-5-methylhexanoic acid, PGB binds with high affinity to the α2δ1 site (a subunit of voltage-gated calcium channels (VGCCs) in the central nervous system [Field et al. 2006]. These high-affinity GBP- and PGB-binding sites are present throughout the dorsal spinal cord and brain. This is a presynaptic channel which modulates release of excitatory neurotransmitters vital for both nociception and epileptogenesis [Taylor et al. 2007]. It is known that gabapentinoids prevent trafficking of the α2δ1 subunit from the dorsal root ganglia neurons to the dorsal spinal cord within animal models of NeP [Bauer et al. 2009]. This α2δ1 subunit binding is thought to be responsible for both antinociceptive and probably its antiseizure effects as well [Vartanian et al. 2006]. Once ligation occurs at the α2δ1 subunit, a reduction in the excessive release of multiple excitatory neurotransmitters occurs; these neurotransmitters include noradrenaline, serotonin, dopamine, glutamate and substance P [Field et al. 2001; Gajraj, 2007; Perret and Luo, 2009]. Finally, PGB may elicit the internalization of VGCC at a cellular level [Weissmann et al. 2013]. PGB’s effect is dependent upon the existence of hyperexcitation of the presynaptic neuron with minimal effects shown to occur during normal neuronal activity [Fink et al. 2002].
PGB is structurally related to the inhibitory neurotransmitter γ-aminobutyric acid (GABA), just as with GBP [Brawek et al. 2009]. In addition to its impact on the α2δ1 subunit, there are suggestions that PGB may also modulate GABA concentrations and the glutamate synthesizing enzyme, branched-chain amino acid transaminase (cytosolic form) [Hutson et al. 1998; Micheva et al. 2006]. GBP may also modulate glutamate synthesis indirectly [Xu et al. 2004] and increase nonsynaptic GABA responses at the GABA-A [Gotz et al. 1993; Lucke et al. 1998] or GABA-B receptors [Parker et al. 2004]. In addition, PGB may enhance activity of the neuronal glutamate transporter type 3, increasing glutamatergic responses [Ryu et al. 2012]. The AED mechanism for gabapentinoids is uncertain, but in animal models, gabapentinoids prevented seizures in rodent models for both maximal electroshock and pentylenetetrazole seizure models [Vartanian et al. 2006]. Finally, another potential mechanism may be gabapentinoid-mediated synaptogenesis [Eroglu et al. 2009] with potential blockade of new synaptic formation. When studied in animal analgesic models, gabapentinoids modulate both hyperalgesia (exaggerated response to a painful stimulus) and allodynia (pain-related behavior in response to a normally innocuous stimulus).
Although differences do not appear to be present between PGB and GBP for mechanisms of action, PGB’s affinity and potency for the α2δ1 subunit of the VGCC is speculated to be higher than that of GBP, although published evidence does not exist. If PGB does have increased VGCC affinity, then this may be the reason why PGB has clinically greater efficacy at lower doses compared with GBP.
After oral administration, PGB is subject to rapid absorption. Oral bioavailability is over 90% and independent of the dose received. This is compared with 30–60% bioavailability for GBP (Table 2). Following either single (25–300 mg) or multiple dose (75–600 mg/day) administrations, there is a linear association for maximum plasma concentrations (Cmax) and area under the plasma concentration–time curve (AUC) values. There is a difference between PGB and GBP for gastrointestinal absorption, although both gabapentinoids are absorbed across the gastrointestinal tract using a system-L transporter system, GBP absorption is solely mediated by this system L transporter, leading to limitation through this saturable, active and dose-dependent transporter, producing nonlinear pharmacokinetics [Bockbrader et al. 2010; Gajraj, 2007]. PGB, however, has nonsaturable absorption, providing linear pharmacokinetics [Bockbrader et al. 2010; Gajraj, 2007]. Both gabapentinoids are also absorbed across the intestinal apical membrane via Na+-independent amino acid transporters [Piyapolrungroj et al. 2001; Su et al. 2005]. However, gabapentinoid transport across the intestinal basolateral membrane is likely mediated by the system L transporter. These factors may also contribute to saturable absorption of GBP across the gastrointestinal tract, as high affinity and lower capacity of GBP saturable transport and its dose-dependent decrease in oral absorption [Bockbrader et al. 2010; Piyapolrungroj et al. 2001; Su et al. 2005]. As such, the rate of PGB absorption is threefold higher than that of GBP. These factors explain how PGB achieves a faster peak blood concentration (1 h post dose) compared with GBP (3 h) [Bockbrader et al. 2010; Gajraj, 2007].
Pregabalin: pharmacokinetics and metabolism with comparison to gabapentin.

This table has been modified from Table 3 in Toth [2012].
PGB has an elimination half life of 5.5–6.7 h, independent of dose and repeated dose administration (Table 2). Elimination of PGB is nearly exclusive to renal excretion, with minimal metabolism at the liver (see below). Renal excretion is supported by data demonstrating that dosing with radiolabeled PGB leads to 90% of the administered dose being recovered unchanged in the urine. There is an N-methylated derivative of PGB, which is a metabolite of PGB found in urine, that accounts for less than 1% of the dose; thus, very little metabolism of PGB occurs in human subjects. Renal elimination occurs at a rate proportional to that of the estimated creatinine clearance (CLCr). Both total and renal PGB clearances are proportionate with CLCr [Randinitis et al. 2003]. Patients with CLCr of 30–60 ml/min are at greater risk of discontinuation due to adverse effects (AEs) than patients having normal CLCr; for this reason, the daily dosing of PGB should be fine tuned for patients with CLCr up to 60 ml/min [Randinitis et al. 2003] and for those patients receiving hemodialysis (Table 2). As mentioned earlier, for patients receiving hemodialysis, a supplemental small dose of PGB could be provided immediately after hemodialysis [Pfizer, 2005; Randinitis et al. 2003] in order to uphold steady-state plasma PGB concentrations. If required, hemodialysis could be used to clear large proportions of PGB [Randinitis et al. 2003].
The oral clearance of PGB is likely to decrease with increasing age; therefore, dose reductions should be considered for older patients. It is best to divide the total daily dose as determined by the dose; for example, if 300 mg/day is targeted, then 150 mg orally twice a day could be provided. If 225 mg/day is suggested, then 75 mg orally three times a day could be prescribed (Table 2).
PGB does not inhibit or induce the major cytochrome P450 system isoenzymes; therefore, PGB is rarely, if ever, associated with hepatic dysfunction. There is only minimal metabolism of PGB at the liver; an N-methylated derivative accounts for an estimated 1% of the dose provided. An absence of hepatic metabolism does not prevent drug-induced hepatotoxicity [Einarsdottir and Bjornsson, 2008], however, as hepatotoxicity due to PGB has been described in isolated case reports [Dogan et al. 2011; Einarsdottir and Bjornsson, 2008; Lindh, 2010; Sendra et al. 2011].
The effects upon anesthesia and the perioperative period are unclear. PGB may possibly be associated with significant respiratory depression postoperatively [Eipe and Penning, 2011]. With PGB now being used more frequently perioperatively for prevention of postoperative pain, this AE may become better defined with experience. Perioperative use of PGB 300 mg provided both 1 h presurgery and 12 h later may contribute to greater AEs, including blurred vision, dizziness and headache compared with patients receiving diazepam 10 mg with a similar dosing schedule [Jokela et al. 2008].
Dosing and initiation of pregabalin
Dosing of PGB can be suited for the individual patient, based on use of other medications, CLCr and their history of prior tolerability to medications. Patients who have a history of developing AEs due to small doses of other medications may have similar reactions to PGB. Starting doses should be 75 mg orally every night at bedtime or 75 mg orally twice a day, with this dose increased gradually as tolerated to a dose of 150 mg orally twice a day over 1–2 weeks based on efficacy. For most patients, PGB is most effective when dosing is optimized at 300 or 600 mg/day, although some patients may do well with lower doses. In general, higher doses of PGB are more likely to be intolerable. If sufficient pain relief is not achieved after 2–4 weeks of treatment using 300–600 mg/day, or if intolerability develops with doses between 75 and 600 mg/day, then these patients should discontinue PGB.
Clinical implications and clinical study outcomes
For patients with painful DPN and PHN, several studies have investigated the potential of PGB for pain relief efficacy and tolerability. For DPN, PGB has been studied through seven randomized, double-blind clinical trials (Table 3). A total of three meta-analyses or pooled analyses have been performed to study the use of PGB for the treatment of DPN [Freeman et al. 2008; Hurley et al. 2008; Quilici et al. 2009]. Doses higher than 150 mg/day are generally suggested for PGB efficacy. This is supported by single studies demonstrating that doses of PGB of up to 150 mg/day are consistently inefficacious [Satoh et al. 2011]; however, a pooled analysis has shown that PGB at doses of 150, 300 or 600 mg/day is significantly better than placebo for patients with DPN [Freeman et al. 2008]. A number needed to treat (NNT) for responders was calculated to be six and four for PGB 300 and 600 mg/day respectively [Freeman et al. 2008]. For this pooled analysis, the onset of sustained improvement in pain had a median time of 4–5 days [Freeman et al. 2008; Sharma et al. 2010].
Important randomized clinical studies of pregabalin for the treatment of NeP conditions.
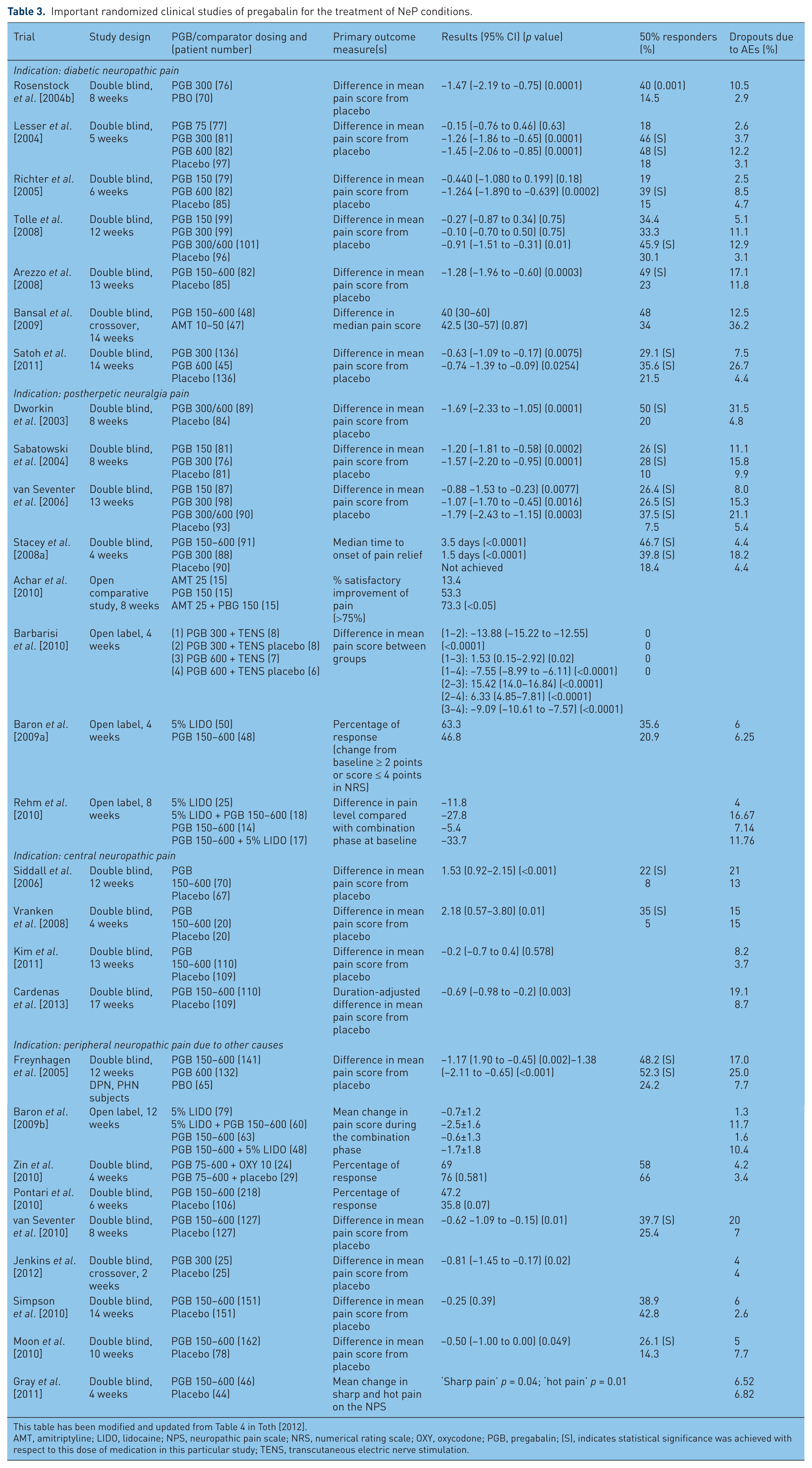
This table has been modified and updated from Table 4 in Toth [2012].
AMT, amitriptyline; LIDO, lidocaine; NPS, neuropathic pain scale; NRS, numerical rating scale; OXY, oxycodone; PGB, pregabalin; (S), indicates statistical significance was achieved with respect to this dose of medication in this particular study; TENS, transcutaneous electric nerve stimulation.
Comparisons of PGB with other NeP agents have been performed. Flexible dosing of PGB at 150–600 mg/day provided greater responders (48% versus 34%), better tolerability and fewer dropouts due to AEs than with amitriptyline, a tricyclic antidepressant (at 10–50 mg/day), but overall efficacy was similar [Bansal et al. 2009]. Comparisons between amitriptyline, duloxetine and PGB have shown similar efficacies in pain relief, with better sleep efficacy but more AEs occurring with PGB compared with the other two agents [Boyle et al. 2012]. As a final point, a recent meta-analysis indirectly compared PGB with duloxetine, a selective serotonergic noradrenergic uptake inhibitor [Quilici et al. 2009], from three studies of duloxetine and six studies evaluating PGB, and found no difference between these two pharmacotherapies for improvement of 24 h pain severity. While PGB was superior to duloxetine for improving the patient’s global impression of change, it led to more dizziness [Quilici et al. 2009]. A recently presented study examined the use of PGB, duloxetine or both in treatment of DPN [Wilhelm et al. 2012]. There did not appear to be any beneficial additive effect of combining these two separately acting pharmacotherapies, while indirect comparisons suggested that duloxetine treatment provided greater average pain relief upon the Brief Pain Inventory average pain outcome measure than PGB. Further comparison studies will be important in future to determine the role of PGB and other potential first-line therapies for the treatment of NeP.
In addition to the large number of studies of patients with DPN, there have been several randomized, controlled trials examining the efficacy of PGB in patients with PHN. A total of four trials have compared PGB at fixed doses of 150, 300 and 600 mg/day with placebo [Dworkin et al. 2003; Sabatowski et al. 2004; Stacey et al. 2008a; van Seventer et al. 2006]. A large retrospective analysis of nine placebo-controlled trials of PGB in patients with DPN or PHN identified patients responding to PGB to achieve this response by the end of only 2 days of treatment [Sharma et al. 2010]. PGB has also been compared with active comparators, including lidocaine 5% topical solution [Baron et al. 2009a], amitriptyline [Achar et al. 2010], transcutaneous electric nerve stimulation [Barbarisi et al. 2010] and 5% topical lidocaine [Rehm et al. 2010] (for each of which PGB was found inferior). For the placebo-controlled trials, all doses of PGB were effective, with responder rates escalating based upon dose: 26% with 150 mg/day of PGB, 26–39% with 300 mg/day and 47–50% with 300–600 mg/day [Dworkin et al. 2003; Sabatowski et al. 2004; Stacey et al. 2008a; van Seventer et al. 2006]. Overall, these results are supported by a meta-analysis of randomized, controlled trials of PGB for acute and chronic pain supports efficacy of PGB for PHN management [Moore et al. 2009].
Low back pain may be the most common cause of chronic pain [Verhaak et al. 1998], affecting 15–45% of the general population [Elliott et al. 1999; Lawrence et al. 1998]. Although often mechanical and nociceptive in nature, neuropathic components are present in 20–35% of this population [Freynhagen and Baron, 2009]. Two randomized, controlled studies evaluated efficacy and tolerability of PGB in patients with low back pain [Baron et al. 2010; Romano et al. 2009], demonstrating both efficacy and tolerability of PGB, the cyclooxygenase inhibitor celecoxib or their combination over 12 weeks of treatment using a double-blind design [Romano et al. 2009]. A double-blind, placebo-substitution study evaluated the time to loss of pain relief response in patients with lumbosacral radiculopathy causing low back pain whose condition had previously responded to PGB using a single-blind, 4-week exposure to PGB [Baron et al. 2010]. However, in the double-blind study phase, PGB and placebo were similar in time to lost response.
Some conditions causing central NeP, pain arising from lesions of the central nervous system, have also been examined for PGB efficacy. These conditions included spinal cord injury, multiple sclerosis or stroke [Finnerup, 2008]. Studies to date have shown that flexible dosing permitted a significantly greater reduction in pain for patients treated with PGB compared with placebo for two of three studies performed (Table 3) [Siddall et al. 2006; Vranken et al. 2008]. Another randomized, placebo-controlled study examining flexible dose PGB for patients with poststroke pain demonstrated no benefits upon pain relief, but PGB improved secondary outcomes, including anxiety, sleep and the clinician’s global impression of change measurement [Kim et al. 2011]. Two studies have appraised pain relief with PGB for pain associated with spinal cord injury, also demonstrating positive efficacy [Cardenas et al. 2013; Siddall et al. 2006].
Post-traumatic NeP is possibly more refractory than other causes of NeP. Studies to date have identified PGB to have pain relief efficacy with good tolerance [Jenkins et al. 2012; van Seventer et al. 2010]. More general studies examining the use of PGB in the management of a variety of NeP conditions including peripheral neuropathy, radiculopathy and trigeminal neuralgia [Navarro et al. 2010, 2011; Perez et al. 2009; Saldana et al. 2010].
As alluded to above, there are some subtle differences between PGB and GBP that may prompt clinical questions about superiority. This question occurs in an absence of any high-quality head-to-head randomized clinical trials examining PGB and GBP. There are some observational studies that have suggested that PGB may have some superior features to GBP [Ifuku et al. 2011; Mishra et al. 2011; Saldana et al. 2012; Tanenberg et al. 2011; Toth, 2010]. A post hoc analysis of two multicenter, prospective, 12-week studies comparing PGB and GBP for patients with DPN, PHN, trigeminal neuralgia and radiculopathy [Perez et al. 2010] showed a greater reduction in the last-week mean pain score and a higher number of responders when PGB was provided. In addition, there were reduced healthcare costs when PGB was used [Perez et al. 2010], and more patients treated with PGB achieved therapeutic dose levels than patients treated with GBP [Gore et al. 2007; Perez et al. 2010]. This may relate to many physicians feeling uncomfortable with GBP dosing, and a lack of understanding about appropriate dosing levels with GBP. For patients with partial epilepsy, a meta-analysis of randomized controlled trials of PGB and GBP found that PGB had improved response rates at doses of 300–600 mg compared with GBP at 1200–1800 mg [Delahoy et al. 2010]. Furthermore, in generalized anxiety disorder patients, benzodiazepine use was reduced more readily in patients receiving PGB as compared to patients receiving GBP [Bramness et al. 2010]. PGB’s use in generalized anxiety disorder [Feltner et al. 2003; Pohl et al. 2005] is also useful to reference when PGB is to be used in patients with NeP and generalized anxiety disorder.
Safety evaluation: adverse effects profile
In most published studies, PGB has been generally well tolerated, both in premarketing clinical studies and with postrelease studies. The majority of AEs experienced are noted to be mild or moderate in severity only. Often, these AEs are transient and present early on at initiation of therapies before later resolution, suggesting that they are self limited. When present after initiation, AEs may dissipate over the first 2–4 weeks of use. Overall, AEs due to PGB are usually tolerated [Hindmarch et al. 2005] and associated with PGB dose received (Table 4). AE profiles with PGB appear to be comparable among all patient populations for incidence; this holds true for sex and for age [Chiechio et al. 2009].
Most common adverse events by treatment group, age, and type of neuropathic pain.
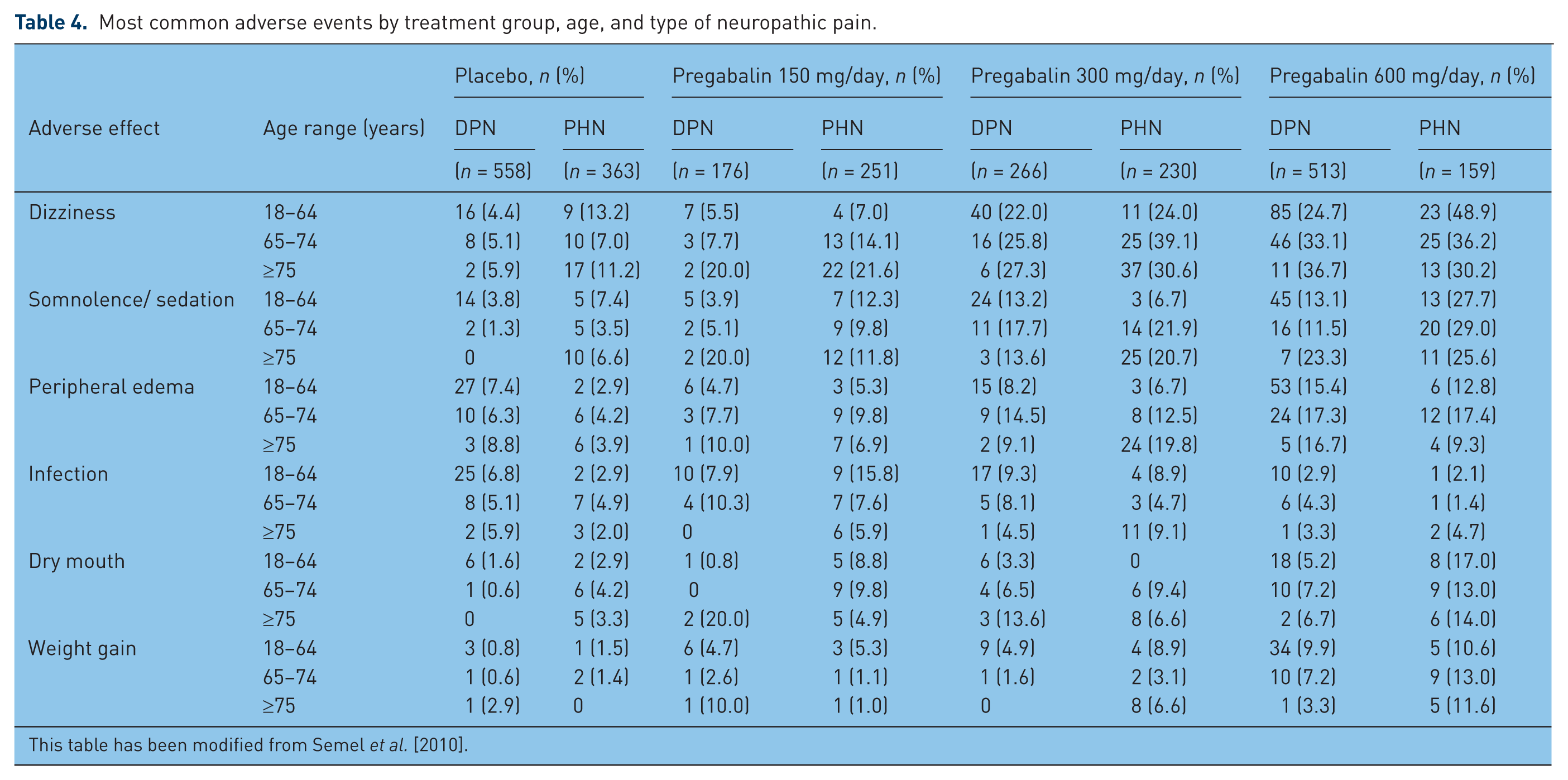
This table has been modified from Semel et al. [2010].
It is unknown whether AEs with PGB differ from those with GBP, as there are no head-to-head comparisons of the two agents. While most of the studies examining GBP featured variable dosing [Rice and Maton, 2001a; Rowbotham et al. 1998], most PGB trials have used fixed dosing without titration. These differences in study designs could impact upon incidences of AEs found in published studies. Despite these differences in trial design, reviewing the available studies demonstrates that AE profiles look quite similar. It is possible that GBP may more frequently lead to nausea and diarrhea [Parke-Davis, 2005], but this is uncertain.
Adverse central nervous system effects
The most common AEs seen among trials of PGB, occurring in at least 10% of any age or dosage group, are dizziness and somnolence (Table 4). The incidence of these most common AEs increases with larger PGB doses. However, this dose-related effect is unlikely to be related to older ages of the dose provided to older age patients. Dizziness and somnolence both arise with moderate frequency; dizziness takes place in 31% of patients treated with PGB compared with 9% of those receiving placebo. Somnolence is slightly less common, being experienced in 22% of patients treated with PGB compared with 7% of those receiving placebo [Pfizer, 2005]. Dizziness and somnolence, alone or together, can impair abilities for performance of potentially dangerous job functions, such as driving or operating complex or heavy machinery. These AEs often occur when PGB is initiated, with these AEs often diminishing after weeks of therapy with PGB. For clinical trials examining DPN or PHN, 9–14% of patients receiving PGB and 4–7% of those receiving placebo discontinued treatment prematurely due to AEs. As would be anticipated based upon their frequency, dizziness (3–4%) and somnolence (2–3%) are the most frequent AEs to lead to drug discontinuation.
When PGB discontinuation is planned, a gradual tapering should occur. An abrupt discontinuation of PGB has uncommonly been linked to development of a syndrome similar to alcohol or benzodiazepine withdrawal. This may also be related to PGB’s purported mechanism of action at GABA [Norton, 2001]. Such withdrawal symptoms can persist for 1–2 days should gabapentinoids be abruptly discontinued. Along with withdrawal from discontinuation, potential for abuse of PGB has been described. This has led to recommendations for caution and monitoring in patients with a history of substance abuse in Europe [Pfizer, 2005]. This is not anticipated by most clinicians, as the gabapentinoids are not considered to be controlled substances in most countries (PGB is a Controlled Schedule V substance in the USA). It is possible that the psychoactive effects of gabapentinoids could contribute to abuse in a very small number of patients, but certainly not in the average patient [Chalabianloo and Schjott, 2009]. Given that euphoria has been described in patients receiving PGB more than anticipated in patients receiving placebo, and that other symptoms such as nervousness, abnormal thinking, depersonalization and amnesia have been described with gabapentinoid use, the clinician must be alert to these uncommon manifestations of PGB use. Therefore, in patients having a prior history of substance abuse (principally for benzodiazepines) [Schwan et al. 2010] or alcoholism, prescription of PGB should be cautiously supervised by the clinician [Schifano et al. 2011].
Other possible AEs affecting the central nervous system include visual blurring, asthenia, euphoria, gait imbalance and cognitive difficulties (primarily with concentration or attention). A greater proportion of patients treated with PGB reported visual blurring (7%) than was reported by patients receiving placebo (2%); in most cases, visual blurring disappears with continued PGB dosing. In the presence of renal impairment, PGB intoxication may lead to encephalopathy associated with triphasic waves on electroencephalography [Lee, 2012].
Adverse systemic effects
Possible systemic AEs include peripheral edema, dry mouth, weight gain, infection, increased appetite and constipation. The cause of peripheral edema due to the gabapentinoids is unknown, but it is relatively common. PGB therapy leads to peripheral edema in 7.6% of patients compared with 0.4% of patients receiving placebo therapy (Table 4). This AE has led to 0.5% of PGB-related discontinuations in clinical trials compared with 0.2% of patients receiving placebo. There does not appear to be any association between peripheral edema and cardiovascular complications (hypertension or congestive heart failure), or with declining renal or hepatic function. Therefore, it is believed that the peripheral edema that occurs appears to be benign, but still intolerable in some patients. Similarly, dry mouth occurs without known pharmacomechanism in 4.2% of patients receiving PGB and appears to be dose dependent; meanwhile, only 0.4% of patients receiving placebo have noted dry mouth.
It is unclear whether PGB treatment is associated with weight gain. For controlled clinical trials conducted examining PGB use for up to 14 weeks, weight gain of at least 7% over weight at baseline was described for 9% of the patients treated with PGB; this was found in only 2% of those receiving placebo [Pfizer, 2005]. However, pooled data have shown that the majority of patients treated with PGB maintain weight within a ±7% range [Cabrera et al. 2012]. If weight gain does occur, it may occur early or late after initiation of PGB [Cabrera et al. 2012]. Despite this potential weight gain in at least some patients receiving PGB, only a handful (0.3%) of the PGB-treated patients withdrew from the study. PGB-associated weight gain appears to be related to dose and duration of PGB exposure; however, it is not associated with baseline values for body mass index, gender or age. Also, weight gain did not occur in an isolated fashion for patients developing peripheral edema. Despite the possibility of weight gain, there is no deterioration of glycemia for patients with diabetes in controlled and longer-term open-label clinical trials [Pfizer, 2005]. In addition, there is a potential interaction with a thiazolidinedione antidiabetic drug; higher frequencies of weight gain and edema were observed in patients taking PGB in combination with these antiglycemic medications.
While infection is listed as an AE in trials assessing PGB [Feltner et al. 2003], the nature of this infection was never elucidated. As a result, these infections were presumed to be viral. It should also be noted that asthenia mimics infectious symptoms and therefore diagnosis of infection may be erroneous.
Although unusual, or even rare, rash as an AE due to gabapentinoid use is possible. The stated incidence of rash with the gabapentoinoids ranges from 0.3% to 1.3% [Arif et al. 2007]. The incidence of rash may be higher in older patients, in whom it was reported to be 5.2% [Rowan et al. 2005]. The most concerning form of rash is a severe purpuric, vesiculobullar rash that may evolve into Stevens–Johnson syndrome. Phase III clinical studies have reported such rash to occur rarely, while a single case of extensive diffuse, erythematous, maculopapular, vesicular, hyperkeratotic and coalescent rash was reported soon after beginning PGB for NeP [Smith et al. 2008]. Other assorted AEs have been described irregularly, including myoclonus [Huppertz et al. 2001], asterixis [Heckmann et al. 2005] and gynecomastia [Malaga and Sanmarti, 2006]. A single case of encephalopathy and corpus callosal edema occurring following abrupt discontinuation of PGB has been described [Oaklander and Buchbinder, 2005; Prilipko et al. 2006]. Finally, there is a single report of rhabdomyolysis occurring in a patient with combined PGB and simvastatin therapy, a known AE with the latter medication [Kaufman and Choy, 2012].
Any risk of neoplasia with PGB is unclear. Preclinical in vivo lifetime carcinogenicity studies of PGB determined unexpectedly high incidences of hemangiosarcoma found in two different mouse strains [Anonymous, 2005]. However, it is uncertain if this is of clinical significance. In clinical trials evaluating differing patient populations with nearly 6400 patient years of experience in patients older than 12 years of age, the presence of newly discovered or worsening pre-existing tumors was found in a total of 57 patients. It is unclear if this is more than would be anticipated at this time [Anonymous, 2005].
Teratogenicity is felt to be of low probability with PGB. As a result, pregnancy category C has been allocated to PGB by the US Food and Drug Administration. Preclinical animal studies have reported an increased incidence of structural abnormalities in fetuses as well as developmental toxicity, including growth hindrance, nervous and reproduction system functional dysfunction and even mortality. As PGB is capable of crossing the placenta, such potential teratogenic effects must be seen as possible. Despite the preclinical data, no controlled data exist for human pregnancies. Therefore, PGB should only be offered to pregnant women in the absence of other options, with the benefits of use outweighing the potential, and theoretical, risks. For men receiving PGB, information of a possible risk for male-mediated teratogenicity should be provided. Preclinical animal studies have demonstrated a higher incidence for specific skull malformations with advanced or retarded ossification, a higher incidence of skeletal abnormalities, lower fetal body mass and visceral malformations in male animals receiving PGB. Also, aged progeny demonstrate neurobehavioral and reproductive dysfunction. Although this may relate synaptic modification to gabapentinoids [Eroglu et al. 2009], this is unclear. PGB is present in animals’ breast milk. We are unclear about the excretion of PGB in human milk. As many medications have excretion within human milk, there is potential for PGB to be present. Therefore, decisions should be made in concert between clinician and patient regarding the need to discontinue nursing or PGB use, with appropriate benefits and risks considered.
Idiosyncratic and hypersensitivity reactions
There are rare descriptions of PGB being associated with hypersensitivity reactions [Smith et al. 2008]. Such AEs include skin blisters, hives, rash, erythema, dyspnea and wheezing. Another described possible reaction is angioedema. This can present with swelling of the face, tongue, lips, gums, throat and larynx, and can be life threatening with respiratory compromise. Should these symptoms occur, just as with any other medication, it is critical that PGB be discontinued immediately. It is possible that other medications capable of inducing angioedema, such as with the angiotensin-converting enzyme inhibitors, may have an increased risk of developing angioedema.
Drug interaction concerns
Interactions between gabapentinoids and other medications are very rare. This is mostly because gabapentinoids do not demonstrate any substantial binding to plasma proteins. Also, PGB does not have induction or inhibitory properties for the major isoenzymes of the cytochrome P450 system in vitro and PGB’s pharmacokinetics are not affected by genetic polymorphisms of cytochrome P450 isoenzymes [Bockbrader et al. 2010]. Coprovision of PGB does not impact upon the pharmacokinetics of GBP, oxycodone, lorazepam, oral contraceptives or ethanol in vivo. For each of these reasons, drug–drug interactions with gabapentinoids are improbable. For other AEDs, with the possible exclusion of tiagabine, there may be negligible pharmacokinetic interactions of clinical insignificance [Bockbrader et al. 2011]. Conversely, there are no particular medications possessing any risk for major interaction with PGB [Cada et al. 2006].
Conclusion
PGB is now a chief consideration for pharmacological management of NeP in DPN, PHN and other conditions. Pain relief efficacy has been shown in NeP conditions, including DPN, PHN, low back pain with or without radiculopathy, post-traumatic NeP, fibromyalgia and for central NeP such as with spinal injury. Its efficacy for pain relief is comparable to other medications used in NeP conditions, and with a benign AE profile. As such, PGB should be considered as a first-line agent beside duloxetine and the tricyclic antidepressants for most patient populations with NeP. When initiating treatment, it is important to start with a low dose of PGB and proceed using slow titration. Twice daily dosing is recommended for maintenance of patient compliance. In patients with renal impairment, the dosing schedule for PGB must be performed with considerations for CLCr and appropriate reduction in dosing. Effective dosing differs from patient to patient, with 300–600 mg daily typically found most efficacious. A lack of drug interactions with PGB makes prescription easy for the nonspecialist clinician. Clinicians and patients should be conscious of the typical AEs, including somnolence, dizziness, peripheral edema and dry mouth. When discontinuation of PGB is needed, it is best to perform slow weaning over several days before stoppage. For situations in which monotherapy is insufficient for NeP management, combination therapy is a consideration. PGB’s mechanism of action should be different than that of the adjuvant, so tricyclic antidepressants, selective serotonergic noradrenergic reuptake inhibitors, another anticonvulsant, an opioid or even a cannabinoid should be considered depending upon the individual patient. Overall, PGB is a valuable and very benign medication to be used in the treatment of NeP. As our knowledge of PGB in different clinical situations increases, it is anticipated that greater confidence will develop for its use.
Footnotes
Funding
Dr Toth received no funding for the creation of this manuscript.
Conflict of interest statement
Dr Toth has received funding for basic science and clinical research from the Canadian Institutes for Health Research, the Alberta Heritage Foundation for Medical Research, Juvenile Diabetes Research Foundation, Pfizer Canada, Valeant Canada, Lilly Canada and Baxter International. He has received speaking honoraria from Pfizer Canada, Valeant Canada and Lilly Canada.