
Abstract
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) have become essential medications in the management of type 2 diabetes mellitus and obesity due to their ability to improve glucose control and facilitate weight loss by enhancing insulin secretion, reducing glucagon release, and slowing gastric emptying. These mechanisms make GLP-1RAs highly effective for metabolic disorders, benefiting patients who require both glycemic control and weight reduction. However, despite their clinical efficacy, GLP-1RAs have been associated with an increased risk of gallbladder disease, including gallstone formation known as cholelithiasis and inflammation of the gallbladder called cholecystitis, especially with prolonged use and higher doses. This review explores the potential mechanisms by which GLP-1RAs may contribute to biliary disease, focusing on the roles of cholecystokinin suppression, bile acid receptor signaling, and alterations in gut-brain pathways. In addition, we present a novel algorithm designed to outline strategies to address the risks of biliary disease in patients treated with GLP-1RAs.
Plain language summary
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) are commonly used to help people with type 2 diabetes and obesity by improving blood sugar control and promoting weight loss. They work by increasing insulin levels, reducing glucagon (a hormone that raises blood sugar) and slowing digestion. Because of these effects, they are very effective in controlling metabolic conditions. However, while these drugs provide significant benefits, they have also been linked to an increased risk of gallbladder problems, including gallstones (cholelithiasis) and inflammation of the gallbladder (cholecystitis), especially when used for a long time or at high doses. This review examines how GLP-1RAs can affect the gallbladder, including changes in digestive hormones, bile acids, and gut-brain signaling. It also presents a new approach to help physicians manage these risks in patients using these drugs.
Introduction
Glucagon-like peptide-1 receptor agonists (GLP-1RAs) have become a potent class of medications used primarily to treat type 2 diabetes mellitus (T2DM) and, more recently, obesity.1,2 These agents mimic the action of endogenous glucagon-like peptide-1 (GLP-1), an incretin hormone derived from the differential processing of proglucagon, which plays a crucial role in glucose homeostasis. 3 GLP-1RAs stimulate glucose-dependent insulin secretion, suppress glucagon release, and slow gastric emptying, which improves postprandial blood glucose levels. 4 In addition, GLP-1RAs have also been found to contribute to weight loss by promoting satiety and reducing appetite, mechanisms that further enhance their therapeutic benefits for people with obesity or T2DM. 5 Given these effects, GLP-1RAs have become a cornerstone in the treatment of metabolic disorders, especially in patients who not only need glycemic control but also weight reduction to improve their overall health outcomes.6–8 The widespread use of GLP-1RAs has raised concerns about their safety profile. Although GLP-1RAs are generally well tolerated, they are associated with gastrointestinal symptoms, pancreatic complications, and occasionally retinopathy, renal events, or injection-site reactions (Table 1).9–16
Main adverse events and complications associated with GLP-1RAs.
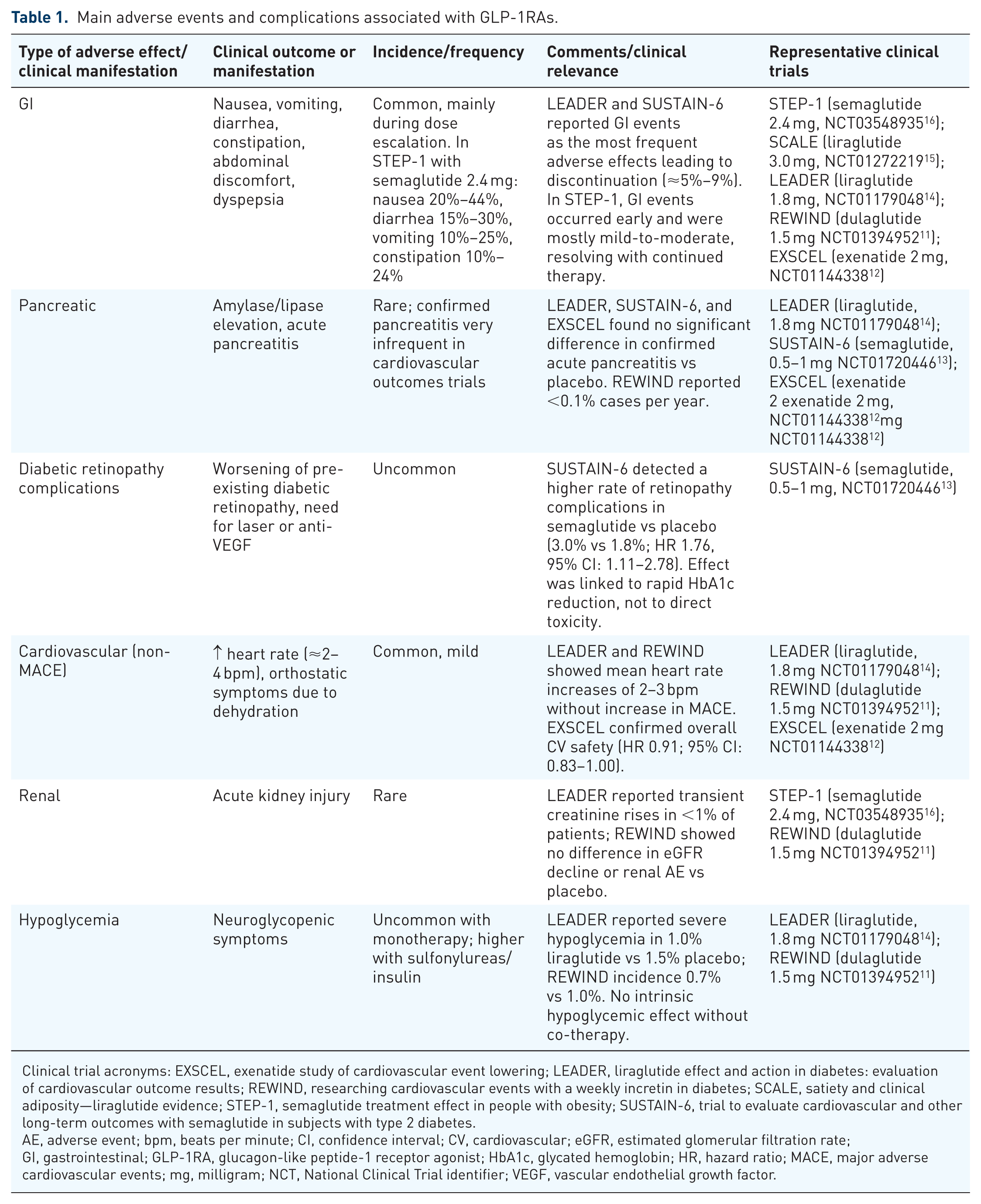
Clinical trial acronyms: EXSCEL, exenatide study of cardiovascular event lowering; LEADER, liraglutide effect and action in diabetes: evaluation of cardiovascular outcome results; REWIND, researching cardiovascular events with a weekly incretin in diabetes; SCALE, satiety and clinical adiposity—liraglutide evidence; STEP-1, semaglutide treatment effect in people with obesity; SUSTAIN-6, trial to evaluate cardiovascular and other long-term outcomes with semaglutide in subjects with type 2 diabetes.
AE, adverse event; bpm, beats per minute; CI, confidence interval; CV, cardiovascular; eGFR, estimated glomerular filtration rate; GI, gastrointestinal; GLP-1RA, glucagon-like peptide-1 receptor agonist; HbA1c, glycated hemoglobin; HR, hazard ratio; MACE, major adverse cardiovascular events; mg, milligram; NCT, National Clinical Trial identifier; VEGF, vascular endothelial growth factor.
Recently, a potentially more serious side effect of GLP-1RAs treatment has received more attention: the increased risk of gallbladder disease, especially in patients with higher doses or prolonged treatment durations. 17 Gallbladder disease, including gallstone formation known as cholelithiasis and inflammation of the gallbladder called cholecystitis, is a major problem, especially in people with obesity and T2DM, the population groups most frequently treated with GLP-1RAs. 18 In these patients, pre-existing risk factors for biliary disease, such as insulin resistance (IR), dyslipidemia, and metabolic alterations, predispose them to abnormal bile composition and an increased risk of gallstone formation. 19 The use of GLP-1RAs may amplify this risk, given its effects on weight loss, gastrointestinal motility, and gallbladder contractility, which could lead to bile stasis and increased gallstone formation.9,20,21 Nevertheless, the mechanisms by which GLP-1RAs may contribute to gallbladder disease have not yet been fully elucidated. The aim of this review is to explore and propose possible mechanisms through which GLP-1RAs may contribute to gallbladder disease and to evaluate the current clinical evidence on the incidence and severity of gallbladder-related adverse events in patients undergoing GLP-1RAs therapy.
GLP-1 RAs: A multifaceted approach to metabolic health
What is GLP-1?
GLP is a peptide hormone produced by the enzymatic processing of proglucagon. Proglucagon is expressed in various tissues, such as the intestine, pancreas, and brain, and undergoes tissue-specific post-translational processing to generate active peptides such as glucagon, GLP-1, and GLP-2. 22 GLP-1 is primarily synthesized in the L cells of the intestinal mucosa, in the α cells of pancreatic islets, and in some neurons of the nucleus of the solitary tract (NTS). 23 Meal intake triggers the release of GLP-1 from L cells in the distal jejunum, ileum, and colon. Upon release, GLP-1 enters the bloodstream and binds to GLP-1 receptors (GLP-1R) located in various tissues (Figure 1). 24 Nevertheless, GLP-1 activity in the bloodstream is short-lived. Under typical physiological conditions, GLP-1 has a half-life of only 1–2 min due to rapid enzymatic degradation by dipeptidyl peptidase IV (DPP-4), which cleaves GLP-1 and renders it biologically inactive.4,25

Systemic effects of GLP-1 receptor agonists on key organs. In the brain, GLP-1RAs reduce food and water intake, inflammation, reward behavior, palatability, and apoptosis, while promoting neuroprotection. In the liver, GLP-1RAs reduce gluconeogenesis and steatosis, while enhancing glycolysis and glycogen synthesis. The gallbladder is affected by decreased motility and contractility, contributing to possible biliary complications. In the heart, GLP-1RAs improve contractility, cardiac output, glucose uptake, vasodilation, and myocyte survival, while promoting glucose utilization. Effects on muscle include increased insulin sensitivity and glucose uptake. In the kidneys, GLP-1RAs promote diuresis and natriuresis, improving renal function. The gastrointestinal tract experiences reduced motility and delayed gastric emptying, which contributes to improved glycemic control and satiety. In the pancreas, GLP-1RAs increase insulin secretion, insulin synthesis, and β-cell proliferation and survival, while reducing glucagon secretion and apoptosis. Finally, in bone, GLP-1RAs stimulate bone formation and increase bone mass. These effects highlight the multifaceted therapeutic potential of GLP-1RAs in the treatment of metabolic disorders, with particular attention to the need to monitor gallbladder-related complications.
Mechanisms of action of GLP-1RAs
GLP-1 RAs are synthetically produced recombinant polypeptides designed to mimic the effects of natural GLP-1, but with a longer duration of action. 26 Unlike natural GLP-1, which is rapidly broken down by DPP-4, GLP-1RAs are structurally modified to resist degradation by DPP-4. This modification allows them to remain active for longer periods, providing a prolonged therapeutic effect. 27
In the pancreas
In pancreatic beta cells, GLP-1 RAs enhance glucose-stimulated insulin secretion by activating the GLP-1R, a G protein-coupled receptor mainly coupled with the Gαs subunit. This activation stimulates adenylate cyclase, which rapidly elevates intracellular levels of cyclic adenosine monophosphate (cAMP). 28 This rapid increase in cAMP activates two key signaling molecules: protein kinase A (PKA) and exchange protein activated by cAMP-2 (Epac2). PKA plays a central role in modulating intracellular processes that enhance insulin synthesis and release. Meanwhile, Epac2 reduces the ATP threshold required to close ATP-sensitive potassium (KATP) channels, which leads to membrane depolarization.29,30 This depolarization opens voltage-gated calcium (Ca²⁺) channels, resulting in a significant influx of Ca2+. The rise in intracellular Ca2+ triggers Ca2+-induced Ca2+ release from intracellular stores, creating a robust signal that primes insulin granules and promotes their exocytosis.31,32 In addition, GLP-1 RAs inhibit glucagon release from pancreatic alpha cells through mechanisms similar to those in beta cells. 33 When GLP-1 binds to its receptor on alpha cells, it triggers intracellular signaling pathways that increase cAMP levels, initiating processes that suppress glucagon synthesis and release. Elevated cAMP activates PKA, which modulates KATP channels by altering their phosphorylation state. This modulation affects the cell membrane potential, leading to a reduction in intracellular calcium levels, an essential factor for glucagon granule exocytosis.4,34
In the gastrointestinal tract
Regarding gastrointestinal effects, GLP-1 RAs bind to GLP-1R located in the stomach and intestines, resulting in modulation of gastrointestinal motility and delayed gastric emptying by binding to GLP-1R on gastric and vagal nerve cells. 35 This interaction triggers a cascade of cAMP-dependent signaling pathways that slow gastric emptying and reduce peristaltic activity in the stomach. The presence of GLP-1R on vagal afferent nerves plays a key role in this effect; when activated, these sensory fibers transmit inhibitory signals to the brainstem, modulating vagal motor output back to the stomach and further reducing motility. This coordinated response causes a delay in the transfer of food from the stomach to the intestines, which not only promotes satiety but also helps regulate postprandial glucose levels by slowing nutrient absorption.36–38
In nervous system
Finally, in the central nervous system, GLP-1RAs regulate appetite and energy balance by binding to GLP-1R in key brain regions, particularly the hypothalamus and brainstem. 39 In the arcuate hypothalamic nucleus, activation of GLP-1 RAs stimulates pro-opiomelanocortin neurons, promoting satiety through α-melanocyte-stimulating hormone and inhibiting hunger-associated neurons expressing neuropeptide Y and agouti-related peptide. 40 This dual action is mediated by intracellular signaling pathways involving AMP-activated protein kinase and mammalian target of rapamycin, which regulate energy balance. In addition, GLP-1RAs act on GLP-1R in the NTS of the brainstem, enhancing vagal afferent feedback from the gut, which reinforces satiety signaling.41,42
Normal physiology of the gallbladder and biliary system
The gallbladder plays a vital role in the digestive process by storing and concentrating bile, a digestive fluid produced by the liver. 43 Bile consists mainly of bile salts, cholesterol, bilirubin, and other compounds that contribute to the emulsification and absorption of dietary fats. Between meals, the gallbladder stores bile, concentrating it by absorbing water and electrolytes. 44 When a meal, especially one rich in fat, is ingested, the presence of fat in the small intestine triggers the release of cholecystokinin (CCK) by enteroendocrine cells. 45 CCK plays a key role in regulating gallbladder function by binding to CCK receptors on the smooth muscle cells of the gallbladder wall, causing gallbladder contraction and the expulsion of concentrated bile through the cystic duct and into the common bile duct, eventually reaching the small intestine. 46 At the same time, CCK also causes relaxation of the sphincter of Oddi, a valve that controls the flow of bile and pancreatic enzymes into the small intestine. 47 In addition to its direct effects on gallbladder contraction and sphincter of Oddi relaxation, CCK is involved in molecular and neuronal pathways that regulate both gallbladder function and satiety. When released in response to dietary fat in the small intestine, CCK binds to CCK1 receptors on vagal afferent neurons, initiating a cascade that activates these neurons and transmits signals to the NTS in the brainstem. This signaling triggers the release of neurotransmitters that induce a feeling of satiety, while coordinating the release of bile as a function of the fat content of the meal.48–50
Traditional mechanisms of gallbladder disease development
Gallbladder diseases, in particular cholelithiasis, are usually due to alterations in bile composition, gallbladder motility, or bile flow regulation. 51 One of the main mechanisms leading to gallstone formation is bile stasis, in which bile remains in the gallbladder for prolonged periods, allowing cholesterol or bilirubin to crystallize and form stones.52,53 Gallstones are usually classified into two main types: cholesterol stones and pigment stones. Cholesterol stones develop when bile becomes saturated with cholesterol, often due to an imbalance in the concentration of bile salts and phospholipids that usually keep cholesterol dissolved. 54 Factors such as obesity, metabolic syndrome, rapid weight loss, and IR contribute to cholesterol stone formation by promoting hypersecretion of cholesterol into the bile and impairing gallbladder motility, which promotes bile stasis and crystallization.55–57 In contrast, pigment stones develop mainly from the crystallization of calcium bilirubinate. This process is usually triggered by an imbalance of bile acids or by bacterial infections, especially in the bile ducts. 58 Infections of the biliary system, where bacterial enzymes hydrolyze bilirubin glucuronides, favor the formation of brown pigment stones. In addition, conditions such as chronic hemolysis, which elevate bilirubin levels in the bile, are common risk factors for pigment stones. 59
Furthermore, cholecystitis, or inflammation of the gallbladder, is a common complication of gallstone disease and usually occurs as a secondary condition to cholelithiasis. 60 When gallstones obstruct the cystic duct, bile flow is impeded, resulting in bile stasis and increased pressure within the gallbladder. This obstruction triggers an inflammatory response in the gallbladder wall, which can cause acute cholecystitis, characterized by severe pain, fever, and tenderness in the right upper abdomen. 61 In addition to mechanical obstruction, bacterial infection can contribute to cholecystitis. Obstructed bile creates an environment prone to bacterial growth, often enteric organisms such as Escherichia coli, Klebsiella, and Enterococcus. These bacteria can enter the gallbladder through the bile ducts or by lymphatic spread, causing an infection that can transform acute inflammation into a more severe form of cholecystitis. 62
The connection between GLP-1RAs and gallbladder disease
Clinical observations have increasingly indicated a possible link between GLP-1RAs use and an elevated risk of gallbladder disease.9,11,13,14,63–69 Recent meta-analyses and systematic reviews have reinforced the association between GLP-1RAs use and an increased risk of biliary and gallbladder disease, especially cholelithiasis.70–72 However, other large-scale observational studies and cohort analyses have not demonstrated a significant increase in risk.73–76 Table 2 summarizes the key studies, their populations, and main findings.
Summary of evidence linking GLP-1RAs to biliary and gallbladder outcomes.

AC, acute cholecystitis; BMI, body mass index; CI, confidence interval; ERCP, endoscopic retrograde cholangiopancreatography; FAERS, FDA Adverse Event Reporting System; GLP-1 RAs, glucagon-like peptide-1 receptor agonists; HR, hazard ratio; NNH, number needed to harm; OR, odds ratio; PS, propensity score; RCT, randomized clinical trial; RD, risk difference; ROR, reporting odds ratio; RR, relative risk; SGLT2i, sodium-glucose cotransporter-2 inhibitor; T2DM, type 2 diabetes mellitus; UDCA, ursodeoxycholic acid.
In a systematic review by He et al., pooling data from 76 randomized controlled trials, it was observed that individuals receiving GLP-1RAs had a 37% higher relative risk of developing gallbladder disease compared with those not receiving them, with a marked increase in cases of cholelithiasis and cholecystitis. This risk was more pronounced in weight-loss scenarios, where higher doses of GLP-1RAs, such as those used with liraglutide and semaglutide, resulted in more significant weight reduction and a correspondingly higher incidence of gallstone-related complications. He et al. 70 emphasized that both dose and duration of treatment with GLP-1RAs play a key role in determining the risk of gallstones, with longer treatment duration and higher doses correlating with a higher likelihood of gallstone formation. In support of these findings, Nreu et al. analyzed 43 trials involving nearly 39,000 patients and reported a 28% increased risk of gallstones among patients receiving GLP-1RAs. Liraglutide, in particular, was associated with an increased incidence of cholelithiasis, whereas other GLP-1RAs, such as exenatide, showed less pronounced associations. Nreu et al. 71 concluded that the increased risk of gallstone formation is not fully explained by weight loss alone, suggesting that the impact of GLP-1RAs on gallbladder motility and bile composition may contribute directly to gallstone risk. Finally, another meta-analysis by Monami et al. focused on GLP-1RAs and safety outcomes, including cholelithiasis; no significant increase in pancreatitis or pancreatic cancer was detected. Nevertheless, the analysis confirmed a 30% increase in the risk of cholelithiasis with the use of GLP-1RAs. Monami et al. 72 noted that this risk remained constant even after adjusting for weight loss, pointing to possible mechanisms beyond weight reduction, such as changes in bile flow or direct effects on gallbladder motility due to chronic GLP-1R stimulation.
These clinical findings underscore the crucial need to explore the underlying mechanisms by which GLP-1RAs may increase gallbladder-related risks. Although weight loss itself is a recognized factor in gallstone formation, it does not fully explain the increased incidence observed with GLP-1RAs use.
Mechanisms by which GLP-1RAs may alter gallbladder function
GLP-1RAs, although beneficial for metabolic control, appear to influence gallbladder function through several mechanisms that may increase the risk of biliary disease (Figure 2). A critical pathway is the suppression of CCK secretion, a hormone necessary for normal gallbladder contraction. Rehfeld et al. have shown that GLP-1RAs reduce postprandial release of CCK, resulting in inadequate emptying of the gallbladder after meals. This impaired contractility contributes to bile stasis, a situation in which bile remains in the gallbladder for prolonged periods, increasing the likelihood of cholesterol crystallization and gallstone formation. In addition, reduced CCK activity affects neural pathways involved in satiety, which could indirectly influence gallbladder motility by altering feedback between the gastrointestinal tract and the brain. 21

Mechanisms linking GLP-1R agonists to gallstone formation. GLP-1RAs suppress CCK secretion, reducing gallbladder contractility and promoting bile retention. They also alter TGR5 signaling, decreasing cAMP-mediated gallbladder relaxation and bile acid-induced GLP-1 secretion. In addition, GLP-1RAs disrupt FXR signaling by reducing FGF19 production, leading to dysregulation of bile acid synthesis and transport through pathways involving cholesterol 7α-hydroxylase (CYP7A1), sterol 12α-hydroxylase (CYP12), and the BSEP. At the neural level, GLP-1RAs activate GLP-1R in the NTS and AP, altering vagal nerve signaling and causing asynchronous contractions of the gallbladder. These combined alterations result in biliary stasis, characterized by prolonged retention of bile and supersaturation with cholesterol, which favors gallstone formation.
At the neural level, GLP-1RAs activate CCK-expressing neurons in the NTS and area postrema, brain regions associated with appetite suppression and nausea. These neurons are activated through GLP-1R signaling, which involves increased cAMP levels and PKA activation that modify the excitability of CCK neurons, potentially leading to altered vagal nerve outflow to the gallbladder. This vagal modulation is critical, as it normally synchronizes gallbladder contractions with digestive needs. Disruption of this neurohormonal feedback loop can result in asynchronous contractions of the gallbladder, promoting prolonged bile retention and further increasing the risk of gallstone formation. 77
The role of FXR in gallbladder function and GLP-1RAs-induced biliary complications
FXR plays a crucial role in gallbladder function, maintaining bile acid balance and regulating motility. 78 FXR is a nuclear receptor activated by bile acids that controls their synthesis and transport by a feedback mechanism. Its activation suppresses bile acid production by inhibiting key enzymes, such as cholesterol 7α-hydroxylase (CYP7A1) and sterol 12α-hydroxylase (CYP8B1). This occurs through two main pathways: (1) FXR induces a small heterodimer partner that represses CYP7A1 and CYP8B1 by inhibiting hepatocyte nuclear factor 4α and liver receptor homolog 1; (2) FXR promotes the intestinal production of fibroblast growth factor 19 (FGF19) and fibroblast growth factor 15 (FGF15), which binds to fibroblast growth factor receptor 4 (FGFR4) in the liver and activates the extracellular signal-regulated kinase and c-Jun N-terminal kinase signaling pathways to further suppress CYP7A1 and CYP8B1.79–83 Regarding transport, FXR modulates key bile acid transporters to maintain proper bile flow and composition. It induces the bile salt export pump to promote the outflow of bile acids from hepatocytes84–86 (Figure 3).

Mechanisms linking GLP-1RAs to gallstone formation through FXR and TGR5. (a) Under physiological conditions, BA activate FXR in hepatocytes and enterocytes, regulating bile acid synthesis by suppressing CYP7A1 and CYP8B1 through induction of SHP. FXR also upregulates BSEP, promoting bile acid secretion. In the intestine, FXR induces FGF19/FGF15, which suppresses BA synthesis in the liver through activation of FGFR4. TGR5, expressed in the gallbladder, is activated by BA, leading to cAMP-mediated smooth muscle contraction, facilitating gallbladder emptying. (b) GLP-1 RAs alter BA homeostasis and gallbladder motility by reducing FXR activation, leading to altered SHP-mediated CYP7A1 suppression, increasing bile acid synthesis, but resulting in an imbalance that favors cholesterol supersaturation in bile. FXR down-regulation also reduces BSEP expression, leading to bile stasis. In addition, GLP-1RAs suppress TGR5 signaling, decreasing cAMP levels and thus reducing gallbladder contractility, further exacerbating bile retention. These changes create an environment that enhances gallstone formation. Dashed red arrows indicate altered pathways under GLP-1RAs treatment, whereas continuous black arrows represent normal physiological regulation.
BA, bile acids; BSEP, bile salt export pump; cAMP, cyclic adenosine monophosphate; FGF, fibroblast growth factor; FXR, farnesoid X receptor; GLP-1RAs, glucagon-like peptide-1 receptor agonists; SHP, small heterodimer associated; TGR5, Takeda G-protein-coupled receptor 5.
Beyond their role in digestion, bile acids also function as signaling molecules that regulate metabolic processes, including the secretion of incretin hormones such as GLP-1. 87 Specifically, activation of Takeda G-protein-coupled receptor 5 (TGR5) by bile acids stimulates GLP-1 release from intestinal L cells. Following food intake, bile acids are released from the gallbladder into the small intestine, where they activate TGR5 receptors on enteroendocrine cells. This interaction triggers the cAMP signaling pathway, which leads to GLP-1 secretion into the bloodstream.88,89 In addition, TGR5 activation also plays a key role in gallbladder motility. When bile acids bind to TGR5 in the smooth muscle cells of the gallbladder, cAMP production increases, leading to muscle relaxation. As a result, bile storage is facilitated, excessive pressure build-up is prevented, and efficient bile flow is ensured.90,91
Furthermore, the interaction between the bile acid receptors FXR and TGR5 adds a layer of complexity to this regulation. While TGR5 activation increases GLP-1 secretion, FXR activation has been shown to exert opposite effects under certain conditions. 92 The regulatory interaction between FXR and TGR5 becomes particularly relevant in the context of treatment with GLP-1RAs. GLP-1RAs alter molecular pathways involving intestinal hormones such as GLP-2 and FGF19, which are essential in bile acid recycling and gallbladder relaxation.93,94 Research by Nerild et al. demonstrated that liraglutide treatment reduces postprandial levels of GLP-2 and FGF19, which normally act through FXR and TGR5 receptors to regulate bile acid homeostasis and gallbladder tone. A reduction of FGF19 and GLP-2 disrupts this receptor signaling, leading to an imbalance between gallbladder contraction and relaxation, thus delaying bile refilling and promoting bile stasis, increasing the risk of gallstone formation due to oversaturated bile. 95 GLP-1RAs can also influence bile acid composition through FXR and TGR5, as described by Gether et al. Reduced FXR and TGR5 stimulation can impair bile acid recycling, resulting in bile that is increasingly oversaturated with cholesterol, which favors gallstone formation. Alterations in FXR and TGR5 signaling not only affect bile composition but also indirectly influence gallbladder motility (Figure 3). 96
The study of cholecystectomy in patients with metabolic dysfunction-associated steatotic liver disease (MASLD) has highlighted the importance of bile acid signaling pathways, including FXR and TGR5, in the maintenance of metabolic and biliary health. Cholecystectomy-induced alterations in bile acid composition and signaling are associated with hepatic steatosis, lipotoxicity, and dysbiosis, which may aggravate the progression of MASLD.97,98 These results underscore the need for further investigation of the interplay between FXR and TGR5, particularly in the context of GLP-1RAs treatment, to better understand the mechanisms underlying biliary and metabolic complications and to guide patient management strategies.
Addressing gallbladder risks in patients on GLP-1RAs
The increased risk of gallbladder disease associated with GLP-1RAs has important clinical implications that require careful consideration in the management of patients undergoing this therapy (Figure 4). 99 Although this association has been documented, current evidence remains limited and inconclusive as to the precise mechanisms and extent of risk. This lack of strong evidence underscores the need for cautious interpretation and individualized patient care. 100 Nevertheless, based on the identification of potential risk factors and emerging data, some practical recommendations can be made to mitigate the risks of gallbladder complications in patients treated with GLP-1RAs.
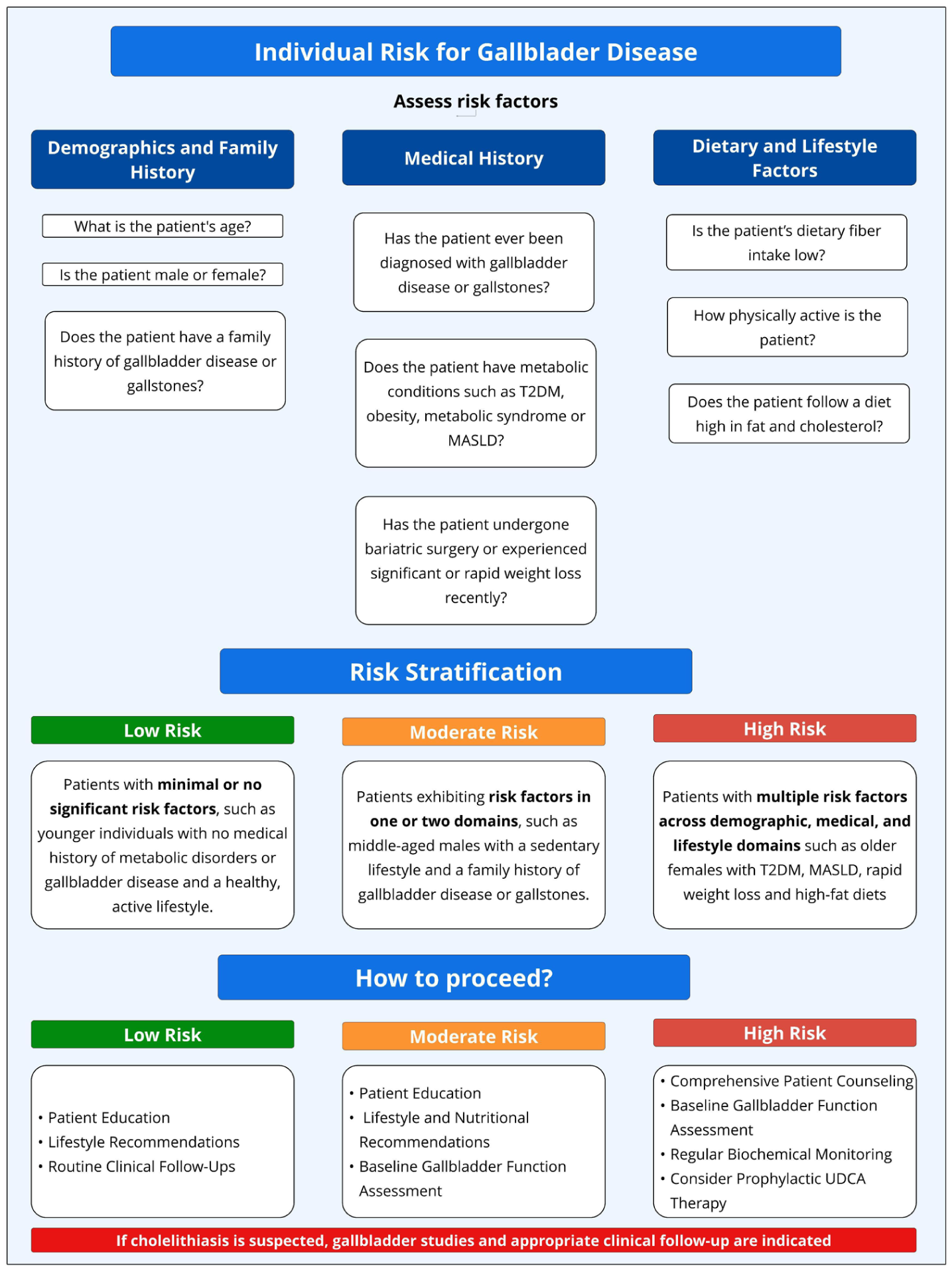
Gallbladder disease risk assessment and stratification in patients undergoing GLP-1RAs treatment. Risk assessment includes demographic (age, sex, family history), medical (history of gallbladder disease, metabolic conditions such as T2DM, MASLD, obesity, insulin resistance, and history of rapid weight loss), and lifestyle factors (diet composition, fiber intake, and level of physical activity). Patients are classified into three risk levels: low risk, which consists of individuals with minimal or no significant risk factors, requiring basic patient education, lifestyle recommendations, and routine clinical follow-ups; moderate risk, which includes those with risk factors in one or two domains, requiring additional lifestyle and nutrition recommendations, patient education, and baseline assessment of gallbladder function; and high risk, which involves individuals with multiple risk factors across demographic, medical, and lifestyle domains, requiring comprehensive patient counseling, baseline gallbladder function assessment, regular biochemical monitoring, and consideration of prophylactic UDCA therapy. Treatment strategies emphasize patient education, lifestyle modifications (gradual weight loss, dietary adjustments, and physical activity), periodic monitoring (clinical, biochemical, and imaging assessments), and personalized treatment approaches to optimize metabolic benefits and minimize gallbladder-related complications. If cholelithiasis is suspected, gallbladder studies and appropriate clinical follow-up are indicated.
Risk factors for gallbladder disease
In addition to the use of GLP-RAs, there are several risk factors that predispose patients to gallbladder disease. Understanding these factors is crucial for the identification and treatment of high-risk individuals undergoing treatment with GLP-1RAs. These risk factors can be broadly classified into demographic, metabolic, pharmacologic, and lifestyle factors. 53 Demographic factors include gender and age, which significantly influence the risk of gallbladder disease. Women are at increased risk due to the effects of estrogen, which increases cholesterol saturation in the bile, thus favoring gallstone formation.101–103 Similarly, advanced age is associated with decreased gallbladder motility and changes in bile composition, predisposing older people to gallbladder-related complications.104,105 Metabolic factors play a major role in the risk of gallbladder disease, with obesity being a major contributor. Obesity increases cholesterol secretion into the bile, leading to oversaturation and an increased likelihood of cholesterol crystal precipitation.106,107 In addition, rapid weight loss, either by strict caloric restriction or bariatric surgery, exacerbates the risk by causing biliary stasis.108,109 IR and hyperinsulinemia, characteristic of T2DM, alter gallbladder motility and bile acid metabolism, further increasing susceptibility to gallstones.110,111 MASLD is another important risk factor, as it shares common metabolic alterations with gallbladder disease, such as dyslipidemia and central obesity, which intensify biliary complications.112,113 Furthermore, lifestyle factors may enhance metabolic risks, exacerbating the likelihood of biliary disease in susceptible individuals. 114 Dietary habits, such as a diet rich in fat and cholesterol, contribute to the supersaturation of bile with cholesterol, accelerating the nucleation of cholesterol crystals and favoring gallstone formation. 115 Conversely, low dietary fiber intake has been associated with impaired bile acid metabolism and reduced gallbladder motility, further increasing the risk of bile stasis.116,117 Physical inactivity is another critical lifestyle factor, as sedentary behavior correlates with slower gallbladder emptying and increased biliary cholesterol secretion, both of which increase the propensity for gallstone development.118,119 Finally, genetic predisposition and family history play a crucial role in the risk of biliary disease. 120 Variations in genes related to bile acid metabolism, cholesterol transport, and gallbladder motility can significantly influence susceptibility. Polymorphisms in genes such as ABCG8 and ABCG5, which encode transporters involved in the flow of cholesterol into the bile, have been linked to an increased risk of gallstone formation. 121
Risk mitigation strategies
Effective management of gallbladder disease risk in patients undergoing GLP-1RAs therapy requires a comprehensive and individualized approach that integrates early risk stratification, patient education, lifestyle modifications, routine clinical follow-up, and targeted therapeutic interventions (Figure 3). 53 Low-risk individuals benefit from general counseling, dietary and lifestyle modifications, and routine follow-up, while moderate-risk patients require additional baseline assessments of gallbladder function. 122 High-risk patients, such as those with multiple predisposing factors like T2DM, MASLD, rapid weight loss, and previous gallbladder disease, need more intensive treatment strategies, including periodic biochemical and imaging follow-up, and consideration of prophylactic treatment with ursodeoxycholic acid to prevent gallstone formation.122–124 Patient education is critical to ensure awareness of biliary complications, emphasizing gradual weight loss, balanced diets, and regular physical activity to optimize bile composition and motility. 125 In symptomatic individuals, timely clinical evaluation, including gallbladder ultrasound and liver function tests, is essential to identify early complications and guide therapeutic decisions. If cholelithiasis is suspected, gallbladder studies and adequate clinical follow-up are indicated to prevent progression to more serious biliary complications.126,127
A multidisciplinary approach integrating endocrinologists, gastroenterologists, and dietitians ensures that the metabolic benefits of GLP-1RAs are maximized while minimizing gallbladder-related complications (Figure 5).

Strategies to address gallbladder disease risks in patients treated with GLP-1RAs. This figure presents a comprehensive approach to mitigate gallbladder disease risks in individuals treated with GLP-1RAs. Initial assessments focus on identifying risk factors, such as gender, history of gallbladder disease, obesity, T2DM, insulin resistance, MASLD, rapid weight loss, dietary habits, lifestyle factors, and concurrent drug use. Imaging techniques such as abdominal ultrasound, when available, are recommended to assess gallbladder health. For high-risk individuals, strategies are divided into actionable categories: (1) Lifestyle recommendations (gradual weight loss, diet modifications, physical activity); (2) Monitoring and follow-up (periodic assessments, diagnostic imaging, and biochemical testing); (3) Personalized treatment (drug dosage adjustments, alternative therapies such as ursodeoxycholic acid or alternative treatments); (5) Understanding risks (knowledge of GLP-1RAs side effects, early symptom reporting, ongoing education); and (6) Information maintenance and involvement (active patient participation in decision making, asking questions, and understanding the risks and benefits of treatment). This framework underscores the importance of having a multidisciplinary team—including endocrinologists, gastroenterologists, hepatologists, and dietitians—to optimize patient outcomes while balancing the benefits and risks of GLP-1RAs treatment.
Conclusion
GLP-1RAs are potent therapies for type 2 diabetes and obesity, but their association with gallbladder disease highlights the need to better understand their effects on bile acid homeostasis and gallbladder function. This review identified key mechanisms, including CCK suppression, disruption of FXR and TGR5 signaling, and alterations in bile acid composition, that may contribute to impaired gallbladder motility and gallstone formation. Current clinical evidence suggests an increased incidence of gallbladder-related adverse events with GLP-1RAs treatment, particularly at higher doses and for longer periods.
These findings underscore the importance of patient risk assessment, follow-up strategies, and tailored interventions to mitigate biliary risks while maintaining therapeutic benefits. Further research is essential to refine our knowledge and guide clinical practice to optimize the safety and efficacy of GLP-1RAs therapy.