
Abstract
SHORT syndrome is a rare genetic multisystemic disorder caused by a loss-of-function mutation in the phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) gene. The disease’s acronym represents its key features: short stature, hyperextensibility, ocular depression, Rieger anomaly, and teeth delay. Insulin resistance, hyperglycemia, and diabetes mellitus are common endocrinological manifestations of this condition. Currently, there are no established guidelines for the treatment of diabetes in SHORT syndrome patients. In this report, we describe a young adult male patient of Chinese descent with atypical diabetes mellitus associated with SHORT syndrome. This case was challenging due to the patient’s young-onset diabetes and poor diabetes control, complicated by insulin resistance from lipodystrophy, and a strong aversion to insulin injections. By utilizing a combination of oral anti-glycemic agents with complementary mechanisms of action (metformin, sodium-glucose co-transporter 2 (SGLT-2) inhibitors, sulfonylureas, thiazolidinediones, and GLP-1 agonists), insulin therapy was delayed. The patient’s blood glucose levels improved significantly, with HbA1c decreased from 14% to 8.8% within 6 months of starting the multi-agent regimen, and further improved to 7.4% with a fasting plasma glucose of 4.8 mmol/L. With an oral medication regimen that the patient found acceptable, both his quality of life and adherence to treatment improved. These findings provide useful insights into tailoring an individualized diabetes treatment plan.
Introduction
SHORT syndrome is a rare autosomal dominant systemic disorder characterized by short stature (S), hyperextensibility (H), ocular depression (O), Rieger anomaly (R), teeth delay (T), and other major anomalies, including intrauterine growth restriction (IUGR), postnatal growth restriction, a distinct facial gestalt (triangular face, hypoplasia of nasal alae with an overhanging columella), insulin resistance, diabetes, and partial lipodystrophy. Less common features include hearing deficits, speech delays, and inguinal hernia. 1 Among these abnormalities, metabolic syndrome, including diabetes and insulin resistance, typically occurs in the second decade of life. The exact prevalence of the disorder remains unclear; likely rarer than 1 in a million, with fewer than 50 cases documented in the medical literature. 1 The underlying cause of SHORT syndrome is a genetic mutation in the phosphoinositide-3-kinase regulatory subunit 1 (PIK3R1) gene (5q13.1), which encodes the critical enzyme phosphatidylinositol 3-kinase regulatory subunit 1. The enzyme is crucial for activating the PI3K/AKT/mTOR pathway—a critical signaling pathway involved in cell growth, proliferation, migration, protein synthesis, transport, cell survival, and adipocyte maturation. This pathway also acts downstream of endocrine hormones, particularly insulin, insulin-like growth factor, growth hormone, and various other growth factors and cytokines. 2 Consequently, loss of function of PIK3R1 leads to insulin resistance, diabetes, and lipodystrophy.
Here, we present a challenging case of young-onset diabetes with a delayed diagnosis of monogenic diabetes, lipodystrophy, and SHORT syndrome due to a genetic variant in the PIK3R1 gene (c.1945C>T, p.Arg649Trp). We have also conducted a comprehensive literature review and developed an individualized treatment approach that significantly improved his glycemic control and treatment adherence. The reporting of this case is in line with the CARE guideline (Supplemental Material). 3
Case presentation
At 21 years of age, a man of Chinese ethnicity was diagnosed with diabetes mellitus from a health screening test with random blood glucose of 11.8 mmol/L, fasting blood glucose of 7.5 mmol/L, and elevated HbA1c of 14%, associated with symptoms of polyuria and polydipsia. Both paternal grandparents and maternal grandmother were diagnosed with type 2 diabetes in their 40s to 60s and were treated with oral hypoglycemic agents. Neither his parents nor his sister had diabetes. There was no other significant family history pertaining to syndromic endocrinopathies, autoimmune conditions, hearing impairment, or growth retardation. Shortly following the diagnosis of diabetes, the patient was referred by a general practitioner to an endocrinology clinic at the age of 22 due to severe hyperglycemia. Following evaluation, the patient was initiated on a multiple daily insulin injection regimen, including glargine (once daily) and glulisine (3 times daily before meals), which was maintained for 2–3 years. Subsequently, to enhance patient adherence, the regimen was adjusted to basal insulin (glargine) once daily for an additional year. However, even with the total daily insulin dose increased from 20 to 82 units, his diabetes control remained suboptimal (Figure 1(a)).

Diabetes control. (a) Trend of HbA1c blood levels with diabetes agents and timeline. (b) A CGM result of the patient was obtained when HbA1c was 8.3% at age 29 years old, but unfortunately, the patient is not willing to have further CGMs because of a localized abscess (c) that was caused by the CGM.
At age 25, the patient was referred to our atypical diabetes endocrinology clinic. A comprehensive medical history and examination were undertaken. The patient was born at 7 months of gestational age to non-consanguineous parents, with his father aged 32 and his mother aged 35 at the time of his birth. He was delivered without complications but was small for gestational age in terms of weight and length. However, his parents and sister had adult heights and weights within the normal range (father: 170 cm, 64 kg; mother: 157 cm, 78 kg; sister: 157 cm, 48 kg). At 1 year of age, he was assessed at a pediatric clinic for postnatal development delay and severe bilateral hearing impairment necessitating the use of bilateral hearing aids thereafter. His electronic medical records indicated that Silver-Russell syndrome was briefly considered by his pediatrician as a potential cause of his short stature, but this diagnosis was never confirmed and was unlikely given his clinical features. He received growth hormone therapy from 2 to 6 years old for his short stature. (2. The patient displayed a triangular-shaped face, large low-set ears, small hands and feet, crooked teeth, and lipodystrophy characterized by muscular limbs and trunk with minimal subcutaneous fat tissue, especially below the gluteal region (Figure 2). There were no signs of acanthosis nigricans or skin tags to suggest severe insulin resistance. An ophthalmology check revealed optic disc cupping.
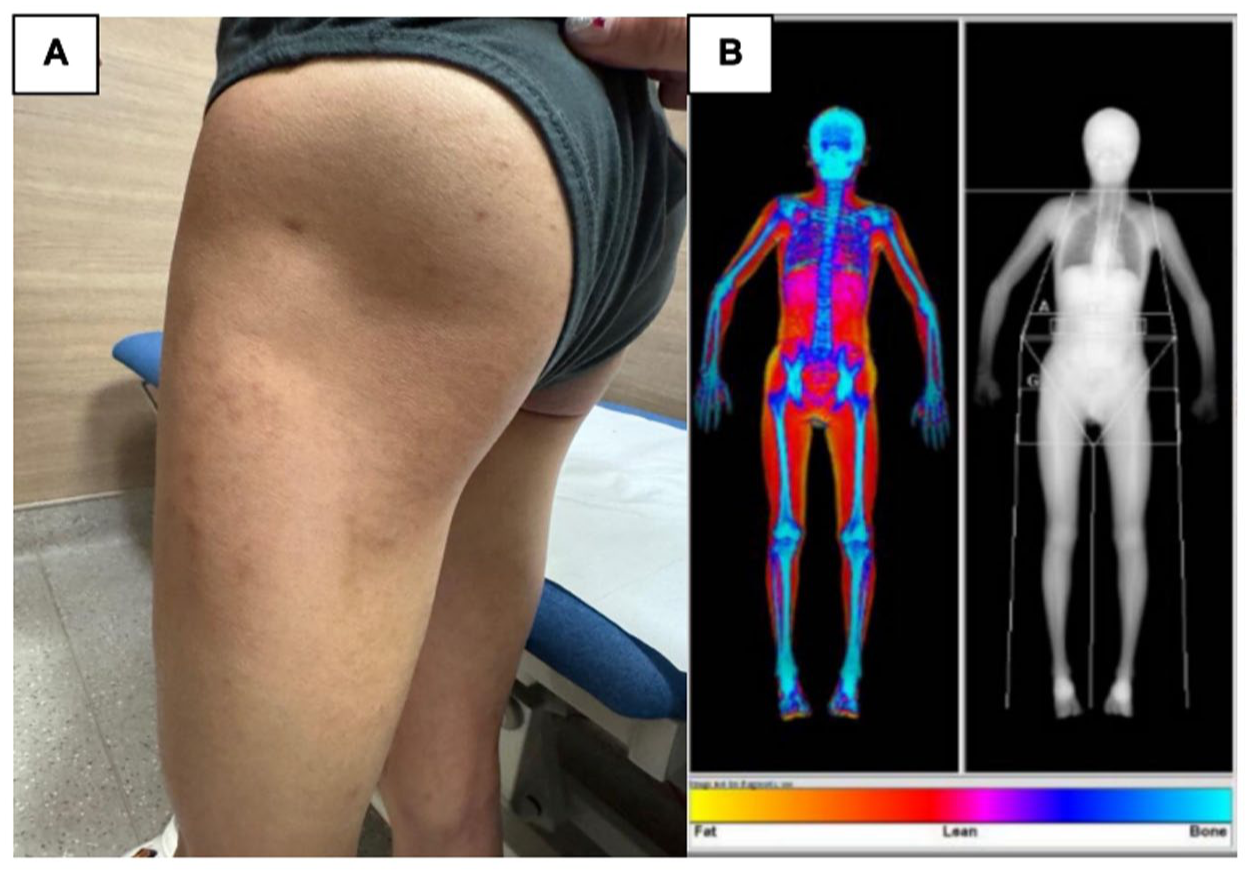
Lipodystrophy on clinical examination. (a) Absence of gluteal fat and subcutaneous fat in the limbs on clinical examination. (b) Generalized reduced subcutaneous adiposity as shown on body composition assessed by DXA.
History from the patient and his parents revealed that he was initially adherent to the regimen of multiple daily insulin injections. However, he stopped being adherent because his HbA1c was consistently suboptimal (above 10%), even at high doses of insulin. The patient expressed frustration with the multiple daily injections required, which affected his work, social life, and overall quality of life. This led to the defaulting of some endocrine clinic visits and strained relationships with family. Due to poor diabetes control, he developed microalbuminuria. He never experienced diabetic ketoacidosis even when he did not take any anti-diabetes medications for several months.
Investigation and management
At the age of 22 years, clinical investigations revealed a detectable C-peptide level of 496.7 pmol/L (deficient when <200 pmol/L) and a markedly increased insulin concentration of 44 mU/L, alongside a fasting blood glucose level of 7.5 mmol/L and an HbA1c level of 14%. These findings suggested that the patient did not have complete insulin deficiency. Instead, he had severe insulin resistance (calculated homeostatic model assessment (HOMA) indice of insulin resistance (HOMA-IR) was 14.7). Negative results for glutamic acid decarboxylase and islet cell antibodies, coupled with preserved C-peptide levels, excluded the clinical diagnosis of type 1 diabetes. 4 Evaluations of the hypothalamic–pituitary–gonadal, hypothalamic–pituitary–adrenal, and thyroid axes were unremarkable. Further investigation of secondary causes revealed a marginally elevated insulin-like growth factor-1 (IGF-1) without clinical features of acromegaly, a normal morning cortisol level (450 nmol/L), and normal liver function. MRI imaging of the pituitary gland and 2D echocardiogram were unremarkable.
Due to the presence of early-onset diabetes mellitus (DM), glaucoma, hearing impairment, short stature, bilateral severe hearing loss since birth, bilateral cataracts, and right eye glaucoma, potential underlying causes of monogenic diabetes involving a systemic pathway were considered. Initial genetic testing of the WFS1 gene, the culprit mutation for suspected Wolfram syndrome (otherwise known as DIDMOAD: diabetes insipidus, diabetes mellitus, optic atrophy, and deafness) in this patient, was performed due to the co-occurrence of diabetes and deafness. However, this diagnosis could not explain certain features of the patient, such as lipodystrophy and the absence of diabetes insipidus. No pathogenic variant was detected. Subsequently, whole-exome sequencing was carried out to explore the underlying genetic causes. DNA libraries were prepared using the Twist Exome 2.0 target enrichment protocol (Twist Bioscience, San Francisco, CA, USA) and sequenced using Illumina NextSeq (Illumina, San Diego, CA, USA) with a read depth of at least 100×. A heterozygous non-synonymous variant (NM_181523.3 (PIK3R1): c.1945C>T p. (Arg649Trp)) was identified as the cause of the patient’s phenotype, and this was confirmed by bi-directional Sanger sequencing (Figure 3). Variants were assessed using the ACMG variant classification guideline (PS2, PS3, PM1, PM2, and PP3) and are classified as pathogenic. The variant is located in exon 15 of the PIK3R1 gene and resides within the Src homology 2 (SH2) domain of the protein product, p85a. Structural modeling predicted that the variant affects protein function by preventing it from interacting with its substrates. 5 This recurrent variant has been previously reported as pathogenic and is known to cause SHORT syndrome (Table 1). 6 Patients with this variant often exhibit typical features of SHORT syndrome, including lipodystrophy and insulin resistance. Targeted Sanger sequencing did not detect the variant in either of the patient’s parents, suggesting that the patient most likely carries a de novo variant.

Genetic sequencing result. (a) Location of the p.(Arg649Trp) variant in the PIK3R1 gene identified in the patient’s DNA. (b) Confirmation of the pathogenic variant by Sanger sequencing. A nucleotide base change from C to T was identified, which resulted in an amino acid residue change from arginine to tryptophan in codon 649. The variant c.1945C>T p.(Arg649Trp) is absent in both parents, indicating a de novo variant.
Literature review of all published reports on SHORT syndrome with diabetes.

S, H, O, R, T individually represent short stature (S), hyperextensibility (H), ocular depression (O), Rieger anomaly (R), and teeth delay (T).
FPG, fasting plasma glucose; IUGR, intrauterine growth restriction; OGTT, oral glucose tolerance test; PCOS, polycystic ovarian syndrome; PIK3R1, phosphoinositide-3-kinase regulatory subunit 1; RBG, random blood glucose; r-HGH, recombinant human growth hormone; s.c., subcutaneous injection; SGLT2, sodium-glucose co-transporter 2; PDA, patent ductus arteriosus; GH, growth hormone; DM, diabetes mellitus; LDL, low-density lipoprotein; OA, osteoarthritis; IR, insulin resistance; N/A, not available; BD, twice daily; FBG, fasting blood glucose; DPP-IV, dipeptidyl peptidase-4.
Considering the patient’s strong reluctance to continue any subcutaneous insulin injections, the patient was restarted on oral medications, including metformin extended release 1000 mg/day (insulin sensitizer), gliclazide 120 mg/day (insulin secretagogue), and pioglitazone 15–30 mg/day (insulin sensitizer) (Figure 1(a)). Despite this, his HbA1c remained high (>10%). Subsequently, daily subcutaneous injections of liraglutide (1.8–3 mg) were initiated and administered for a duration of 1 year, resulting in a notable improvement in HbA1c levels to 9%. The patient was also started on empagliflozin (25 mg). Liraglutide daily injection was stopped at the patient’s request and changed to oral glucagon-like peptide-1 receptor agonist (GLP-1RA) semaglutide (14 mg). The dose escalation was gradual over weeks for liraglutide to the maximum dose of 3 mg with nil major gastrointestinal side effects, but this was stopped at the patient’s request as he did not require any injectable therapies. Similarly, monthly titration of dose for oral semaglutide to the maximum dose of 14 mg, with good tolerability. Over the treatment duration, his weight was stable between 34.3 and 36 kg. This regimen significantly enhanced his diabetes management, resulting in an HbA1c level between 7.4% and 8.3%. With improved diabetes control and the use of an angiotensin receptor blocker (ARB) and empagliflozin, his urine albumin–creatinine ratio improved, declining from 4.1 to 0.7 mg/mmol.
A recent 11-day continuous glucose monitoring using Dexcom revealed an average glucose of 12.5 mmol/L, a predicted HbA1c of 8.7%, and postprandial hyperglycemia (Figure 1(b)). Based on these findings, the gliclazide dose was increased from 60 to 90 mg/day, with home glucose monitoring aimed at maintaining glucose levels between 4 and 10 mmol/L. The patient declined further continuous glucose monitoring due to a localized abscess that developed after the initial use but agreed to continue home capillary glucose monitoring (Figure 1(b)). As his mean glucose readings remained high throughout the day, he would benefit from basal insulin, but he has declined for now, but agrees that he would need to restart basal insulin in the future.
Discussion
We present a rare and unique case of syndromic young-onset diabetes mellitus, specifically the multi-systemic SHORT syndrome, caused by a de novo heterozygous p.(Arg649Trp) variant in PIK3R1. Our patient exhibited classical features of the syndrome, including short stature, ocular depression, delayed dentition, IUGR, partial lipodystrophy, and insulin resistance. Notably, the patient’s diabetes was not well controlled with multiple daily insulin injections, at least not sustainably, due to both a reluctance to continue insulin therapy and the underlying insulin resistance. The patient was managed with an oral regimen that included metformin, a sulfonylurea, a PPAR-γ-agonist, a GLP-1RA, and a SGLT2 inhibitor. This case contributes to the limited literature on the successful use of GLP-1RA and SGLT2 inhibitors in managing diabetes in patients with SHORT syndrome. 16
To date, PIK3R1 is the only gene known to be responsible for SHORT syndrome. A literature review revealed that the genetic variant identified in our patient, p.(Arg649Trp), is one of the most common variants among all known genetic variants associated with this condition, with significant phenotypic heterogeneity (Table 1). 24 Some cases with the same variant exhibited additional features such as permanent ductus arteriosus, polycystic ovaries, hearing impairment, and failure to thrive (Table 1). 25 Several other cases of SHORT syndrome with the p.(Arg649Trp) variant have shown signs of insulin resistance, with affected individuals ranging from neonates to young adults. Notably, insulin-resistant diabetes mellitus is mostly diagnosed during late adolescence to young adulthood. It has been suggested that puberty-related endocrinological changes, rather than the use of growth hormone during early childhood due to growth delay, play a role in the development of insulin resistance and the onset of diabetes. 16 A case series of four Japanese individuals with SHORT syndrome reported a common history of intrauterine growth retardation and young-onset diabetes, due to the disruption of binding of p85α (regulatory subunit of P13K), thus impairing insulin signaling pathway. 23 Diagnostic delays of this rare condition appear to be common, such as in our case, and a case report with a PIK3R1 variant (c.1957A>T) identified 19 years after diabetes onset 22 (Table 1).
PIK3R1 encodes a regulatory subunit of the phosphoinositide 3-kinase (PI3K), an enzyme that plays a key role in regulating cellular processes, including growth, survival, metabolism, and trafficking. 26 PIK3R1 interacts with insulin receptor substrates (IRS) and growth factor receptors, and congenital defects in PIK3R1 result in the dysregulation of the PI3K pathway. This dysregulation of PI3K signaling pathway contributes to growth retardation and insulin resistance in most individuals with SHORT syndrome. In addition to SHORT syndrome, other rare genetic conditions can present with a combination of growth impairment, metabolic dysfunction, and severe insulin resistance, such as SOFT syndrome (Short stature-Onychodysplasia-Facial dysmorphism-hypoTrichosis), a monogenic ciliopathy caused by POC1A variants. 27 Other rare syndromes include Berardinelli-Seip congenital lipodystrophy (BSCL), Hutchinson-Gilford progeria syndrome (HGPS), 28 and SHORT-like syndrome (PTPN11-related). 29
Unlike the more commonly recognized forms of diabetes, such as type 1, type 2, and gestational diabetes, there is limited clear guidance on the management of rare forms of insulin-resistant diabetes, including lipodystrophies. Therefore, individualized treatment plans that consider the underlying pathophysiology are crucial, while genetic testing and body composition scans may help provide insights into personalized therapy.30,31 Lessons for diabetes management can also be drawn from the use of anti-cancer PI3K inhibitors such as alpelisib in breast cancer. Dysregulated PI3K signaling is observed in cancers such as breast cancer. Thus, PI3K inhibitors in cancer treatment aim to reduce the hyperactivation of PI3K signaling in cancer cells. Nonetheless, the inhibition of the PI3K pathway results in a prevalent side effect: hyperglycemia has been reported in 63.7% of patients undergoing treatment with alpelisib–fulvestrant, and diabetic ketoacidosis occurs in 0.7% of patients, attributable to the inhibition of PI3K signaling in skeletal muscle and the liver. 32 For PI3K inhibitor-induced hyperglycemia, suggested treatment includes metformin, possibly combined with thiazolidinediones or a DPP-4 inhibitor, with insulin used as a rescue medication.33,34 However, further workup is necessary to elucidate the ideal diabetes regimen for PI3K inhibitor-induced hyperglycemia and SHORT syndrome.
From our literature review shown in Table 1, there have been several case reports that found metformin,8,14–17,19,20 thiazolidinediones,11,15,17 and sulfonylureas,8,17 and sometimes with high doses of insulin, useful to manage diabetes in patients with SHORT syndrome. Metformin, an effective insulin sensitizer, has been reported to help three teenagers with SHORT syndrome diabetes.14,20 However, there have also been reports suggesting that metformin may worsen insulin resistance and increase insulin concentrations on oral glucose tolerance tests after initiation, although the reasons for this are unclear.13,14 Thiazolidinediones such as pioglitazone taken with metformin, improved a patient’s BMI, insulin resistance, HbA1c, and hyperinsulinemia. 15 The combination treatment of gliclazide, with metformin and pioglitazone, improved HbA1c from 9.1% to 7.1% in a 23-year-old female patient. 17 A recent case report also found that metformin and α-d-glucosidase inhibitor (voglibose) improved glucose and postprandial insulin blood levels but increased fasting insulin levels in a Chinese female patient. 21
In recent years, there have been several reports on the use of SGLT2 inhibitors in patients with SHORT syndrome. A case report showed that a combination of SGLT2 inhibitor with metformin improved HbA1c without episodes of hypoglycemia. 16 Of note, the recently published EMPIRE trial was a single-arm open-labeled Japanese studies that evaluated the safety and efficacy of use of empagliflozin in patients with rare insulin resistance syndromes including lipodystrophy (three of whom had PIK3R1 variant), with a mean BMI of 22.3 kg/m2.35,36 The use of empagliflozin reduced mean HbA1c level by 0.99% points and mean fasting glucose by 3.55 mmol/L, with no serious adverse events including diabetic ketoacidosis, and no clinically significant weight loss (mean −1.68 kg) up to a duration 1-year follow-up. Insights from patients with other forms of insulin resistance and generalized lipodystrophy suggest that GLP-1RA and dual GIP (gastric inhibitory polypeptide)/GLP-1 agonists, such as tirzepatide, could be effective in reducing basal insulinemia and improving insulin resistance.37,38 There is a paucity of data related to the use of sulfonylurea in patients with SHORT syndrome, such as a case report in 1994 8 (Table 1).
Our case suggests that a combination of metformin, thiazolidinediones, sulfonylureas, SGLT2 inhibitors, and GLP-1 agonists may offer a safe and reasonably effective regimen for managing severe hyperglycemia in patients with SHORT syndrome in a young man, although more studies are required to evaluate long-term efficacy and safety of this multi-oral therapy approach. Based on the timing of medication administration, GLP-1RA and possibly SGLT2 inhibitors appear to be the most critical components, providing additional benefits such as cardiovascular protection. 39 In conclusion, we present a case of challenging diabetes management that transitioned from multiple daily insulin injections to a combination of oral diabetes medications with complementary mechanisms of action, improving both diabetes control and patient adherence. The use of precision medicine, including whole-exome sequencing, played a crucial role in diagnosing this de novo case of SHORT syndrome, as well as providing valuable insights for prognosis and tailoring an individualized diabetes treatment plan.
Patient perspective
The patient and his family have been supportive and compliant with his treatment plan. The diagnosis of his condition helped the patient and his parents come to terms with the multiple, previously unexplained health issues he had been experiencing since he was born. Previously, he struggled with the inconvenience of insulin injections and eventually stopped using insulin entirely. Since he never experienced diabetic ketoacidosis or severe symptoms, he found it difficult to convince himself to continue with the multiple daily insulin injections. However, since starting an oral medication regimen taken only in the morning (once/day), his adherence has significantly improved, and he has been able to continue working full-time without major difficulties. He was aware that insulin treatment would eventually be necessary, likely in the near future.
Supplemental Material
sj-docx-1-tae-10.1177_20420188251405363 – Supplemental material for Diabetes mellitus in SHORT syndrome managed with multi-agent oral therapies: a case report and literature review
Supplemental material, sj-docx-1-tae-10.1177_20420188251405363 for Diabetes mellitus in SHORT syndrome managed with multi-agent oral therapies: a case report and literature review by Yufan Yang, Si Hua Clara Tan, Su Chi Lim and Wann Jia Loh in Therapeutic Advances in Endocrinology and Metabolism
Footnotes
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.