
Abstract
Hormonal induction of spermatogenesis offers men with azoospermia due to hypogonadotrophic hypogonadism (HH) the promising prospect of fertility restoration. However, an important exception is the subset of individuals affected by congenital hypogonadotrophic hypogonadism (CHH), also known as Kallmann syndrome if associated with anosmia, who often display dismal responses to fertility induction, despite prolonged therapy. This primarily stems from the loss of minipuberty, which is a crucial phase of testicular maturation in early life that has a far-reaching impact on eventual spermatogenic capacity. Further exacerbating the compromised reproductive health is the failure to initiate timely pubertal induction in many CHH patients, resulting in suboptimal genital and psychosexual development. In this paper, the clinical implications and management of male HH across the lifespan is comprehensively reviewed, with a special focus on novel strategies that have the potential to modify disease severity and maximize fertility potential in CHH by addressing the inadequacies of conventional approaches.
Keywords
Introduction
Hypogonadotrophic hypogonadism (HH) is defined as a clinical syndrome of gonadal failure due to deficient hypothalamic gonadotrophin-releasing hormone (GnRH) or pituitary gonadotrophin secretion. Whereas men with primary testicular dysfunction have irreversibly diminished spermatogenic capacity, gonadotrophin therapy provides men with HH the opportunity to restore endogenous testosterone production and spermatogenesis. Indeed, the prospect of fertility can be greatly enhanced by appropriate spermatogenesis induction treatment, backed by modern assisted reproduction technology in less robust responders, or those with less fertile partners. However, it has been long recognized that a significant proportion of men with congenital forms of HH do not respond favourably to conventional treatment approaches and, in this article, we explore possible reasons for this. Insights into the ontogenesis of the male reproductive tract and accumulating clinical evidence now support novel strategies in which interventions at various stages of life in individuals with more severe disease can potentially improve long-term sexual and reproductive outcomes.
Triphasic testicular maturation and hormonal control of spermatogenesis
In contrast with females who are born with their lifetime supply of oocytes, males require three distinct phases of testicular maturation to develop and sustain spermatogenesis. The first phase of testicular development commences in utero at week 7 of embryonic development, with the secretion of testosterone by foetal-type Leydig cells under the regulation of placental human chorionic gonadotrophin (hCG).
This is followed by ‘minipuberty’, a transient activation of the hypothalamic–pituitary–testicular (HPT) axis during the third trimester of gestation that continues until 6 months postnatal. 1 A hallmark of this developmental phase is the robust gonadotrophin-releasing hormone (GnRH) activity, accompanied by the rise of serum testosterone and gonadotrophin levels to near-adult male levels at 2–3 months of age. 2 During this period, inguinoscrotal testicular descent and anchoring in the scrotum, as well as penile growth, are driven by luteinizing hormone (LH)-induced testosterone and insulin-like 3.3,4 Concurrently, active proliferation of Sertoli cells occurs under the stimulation of follicle-stimulating hormone (FSH), marked by exponential increase in serum inhibin B (IB) and anti-Müllerian hormone. 2 As androgen receptors are not expressed on Sertoli cells until 5 years of age, germ cell maturation and spermatogenesis do not occur. 5 Thereafter, the decline of reproductive hormones to low levels signifies the end of minipuberty, and the HPT axis retreats into quiescence for the rest of childhood.
The reactivation of pulsatile GnRH secretion in early adolescence heralds the onset of puberty, and is detectable clinically by testicular enlargement of ⩾4 ml. Another wave of FSH-stimulated Sertoli cells and spermatogonial proliferation sets in, with a resultant increase in the length and diameter of the seminiferous tubules. They eventually account for 90% of the entire testicular volume (TV), representing key determinants of spermatogenic potential. 6
Subsequently, under the influence of Leydig cell-derived testosterone, Sertoli cells cease to proliferate further and undergo maturation to establish the microenvironment necessary to facilitate spermatogenesis. Sperm production is now possible under the concerted actions of FSH and intra-testicular testosterone. 7 Importantly, the high local concentration of testosterone, which is 40-fold greater than the systemic levels, 8 is a prerequisite for its paracrine effects on the stimulation of spermatogenesis.
Causes of hypogonadotrophic hypogonadism
Differentiating HH into congenital and acquired aetiologies has important clinical implications. First, congenital causes are uncommon but tend to have a more significant impact on reproductive function because of the early disruption to gonadal development. Second, acquired causes may either be permanent or medically reversible, requiring very different management approaches, hence necessitating accurate elucidation of underlying pathology.
Congenital
Congenital hypogonadotrophic hypogonadism (CHH) is a rare genetic condition characterized by deficiency in secretion or action of GnRH, with otherwise normal pituitary function. From limited published literature, a prevalence of 1 in 4000–15,000 is currently estimated.9,10 Although a marked male preponderance (~4:1) 11 in CHH has been consistently reported, it remains incompletely explained at the genetic level, as only a small fraction is identified to be of X-linked inheritance (Figure 1).
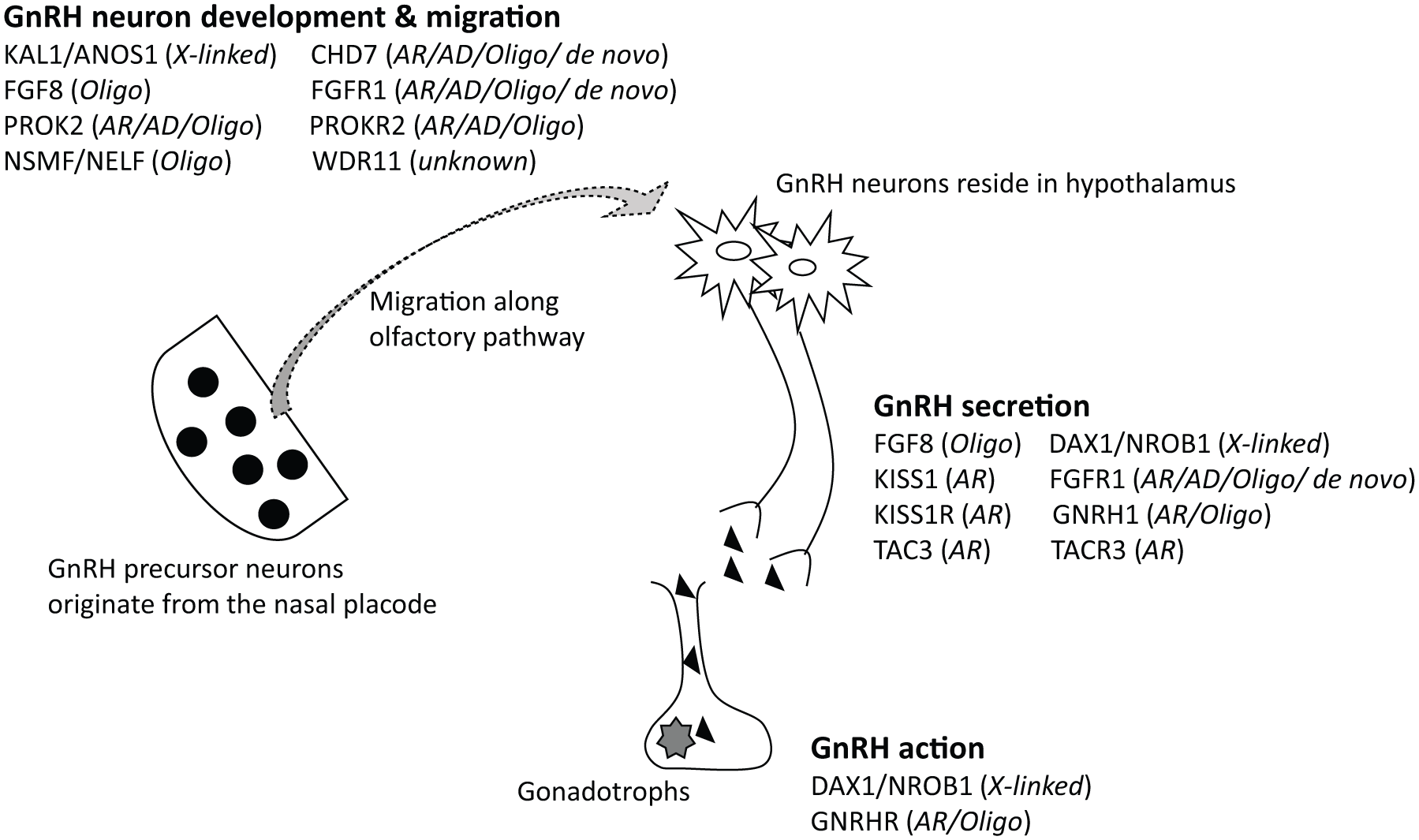
Key genetic mutations involved in the pathogenesis of CHH, with corresponding mode of inheritance. 12
Broadly speaking, the genetic defects underlying CHH can be classified into two main groups: abnormal GnRH and olfactory neuronal migration (anosmia/hyposmia, that is Kallmann syndrome), and impaired GnRH secretion or action (normosmic CHH). This simple dichotomy provides a convenient way to categorize patients clinically based on their sense of smell, but belies the diversity of the genetic mutations involved; over 30 gene loci have been implicated to date and half of the CHH cases remained unaccounted for. 12 This complexity is also reflected in the various modes of transmission possible, including all forms of classical Mendelian inheritance and oligogenicity.12,13 (Figure 1). Additionally, CHH is phenotypically heterogeneous, with variable association with other nonreproductive abnormalities such as cleft lip and palate, dental agenesis, ear defects, deafness, renal agenesis, bimanual synkinesis (mirror hand movements) and skeletal anomalies. 13
Multiple pituitary hormonal deficiencies can occur as a result of defective development and organogenesis of pituitary gland, a condition known as combined pituitary hormone deficiency (CPHD). With a prevalence of 1 in 8000, it is defined by the impaired production of growth hormone, resulting in growth retardation and failure to thrive in children, along with a deficiency of one or more other anterior pituitary hormones, most commonly LH and FSH.14,15 Pituitary hormone deficiencies may develop over a variable period of time during life and are not necessarily all present neonatally. Interestingly, overlapping genetic overlap has been observed between CHH and CPHD with HH, but most individuals are presently of an unknown genotype.14,16 Unlike CHH, the pituitary gland is usually of abnormal appearance in CPHD and isolated growth hormone (GH) deficiency.
Acquired
Organic causes of HH typically result in permanent hypogonadism and are commonly associated with other anterior pituitary hormonal deficiencies. They include destructive and infiltrative disorders of the hypothalamus or pituitary gland, such as pituitary adenomas, craniopharyngiomas, sarcoidosis, haemochromatosis, lymphocytic hypophysitis and histiocytosis, as well as iatrogenic causes including pituitary surgery and radiation therapy. Of note, haemochromatosis preferentially affects gonadotrophs due to the expression of an iron transporter in their cell membranes, and thus tends to manifest as isolated HH (± diabetes, arthralgia, cirrhosis, or cardiomyopathy). 17
In contrast, functional causes of HH, leading to suppression of gonadotrophin and testosterone production, are potentially reversible by disease remission or medical treatment, or discontinuation of the offending medication. One of the most frequent causes of acquired isolated HH is hyperprolactinaemia, which can result from the use of medications that interfere with the dopaminergic system (especially antipsychotic agents), prolactinomas, and less frequently pituitary stalk damage. Other drugs that can reversibly suppress the HPT axis include opiates, glucocorticoids and androgens or anabolic steroids (AASs).
In addition, nutritional disorders (low levels of leptin 18 ) or any undiagnosed chronic illness 19 may also negatively affect hypothalamic GnRH secretion, as is the case in women with hypothalamic amenorrhoea. Obesity-induced HH is being increasingly described in men (but not women), and is postulated to reside in adipose tissue-derived factors, including proinflammatory cytokines and altered insulin signalling, exerting central inhibitory effects on the HPT axis.20,21 However, it is unclear whether these men are genuinely hypogonadal, or just have a form of non-gonadal illness. Moreover, obese men having insulin resistance and low levels of sex hormone-binding globulin (SHBG), may still have physiological levels of free testosterone even when levels of total testosterone are low. 22
Clinical manifestations and diagnostic evaluation
Infants
Because of the lack of minipuberty, a high proportion of male infants with congenital GnRH or gonadotrophin deficiencies are affected by cryptorchidism or micropenis. In the largest United Kingdom (UK) series of men with CHH, around 50% were cryptorchid at birth, of which one-third had bilateral disease, 23 compared with the UK birth prevalence of 5–6%,24,25 whereas micropenis affects 4.6–25.4%26–28 of CHH males compared with the general population birth prevalence of 0.015–0.35%.29,30 Experienced parents may also observe the absence of erections on nappy change. In addition, children may manifest nonreproductive CHH-associated phenotypes at birth, including cleft lip or palate, syndactyly or other anomaly of digits, and hearing impairment detected on automated otoacoustic emissions test. 31 However, anosmia usually cannot be reliably detected until after age 6–8 years.
In contrast with the diagnostic challenges of CHH in early adolescence (discussed later), the phase of male minipuberty provides an extraordinary window of opportunity in which the diagnosis can be established in infancy. By biochemical profiling of reproductive hormones during this period, ideally during the physiological peak of HPT axis activity expected around 1–3 months of age, the findings of abnormally low testosterone, FSH and LH levels would be confirmatory for CHH. Such a strategy was proven to be useful in identifying concomitant HH in a cohort of male infants with congenital hypopituitarism. 32 Moreover, there is also a possibility of thereby inducing testicular descent without surgery, although evidence is sparse. Therefore, proactive screening for absent minipuberty in infants with ‘red flag’ features (Table 1), particularly bilateral cryptorchidism or micropenis, is advocated. 31
Suggested approach to the management of CHH in males.

AMH, anti-Müllerian hormone; BMD, bone mineral density; CDGP, constitutional delay in growth and puberty; CHH, congenital hypogonadotrophic hypogonadism; E2, oestradiol; FBC, full blood count; FSH, follicle-stimulating hormone; hCG, human chorionic gonadotrophin; IB, inhibin B; IM, intramuscular; LH, luteinizing hormone; MRI, magnetic resonance imaging; PSA, prostate-specific antigen; SHBG, sex hormone-binding globulin; T, testosterone.
Red flags: 31 cryptorchidism, micropenis, absent erections on nappy change, anosmia/hyposmia, clefting, digit anomaly, deafness, family history.
Adolescents
Failure to develop secondary sexual characteristics due to pubertal delay is the main mode of presentation in adolescents with HH. In boys, the first sign of puberty is testicular enlargement, which normally occurs between 9 and 14 years old, corresponding to 2–2.5 standard deviations of the population mean. 33 Accordingly, delayed puberty is defined as the absence of testicular enlargement (<4 ml) by age 14 years.
During adolescence, the single most common cause of delayed puberty is constitutional delay in growth and puberty (CDGP), which accounts for 65–82% of affected boys.34,35 Although CDGP represents a late variant in the normal spectrum of pubertal timing, it can only be diagnosed after the exclusion of pathological causes. Therefore, careful clinical assessment in conjunction with biochemical evaluation, such as full blood count, renal and liver profile, inflammatory markers, thyroid function and iron studies, is imperative to rule out an underlying chronic disorder. 36 In addition, pituitary function should be screened by measuring basal hormonal levels. Further investigations, such as dynamic endocrine function tests (rarely necessary) and cranial imaging, should be guided by baseline clinical and laboratory findings.
Notably, CDGP and CHH are difficult to distinguish in teenage years as isolated HH is common to both conditions, although linear growth deceleration is rarely a feature of CHH. A variety of physiological and stimulation tests have been proposed over the years, with some promising results emerging from recent studies on serum inhibin B, but there is yet a ‘gold standard’ test that can reliably differentiate them apart. 37 Genetics may have a role in future as CDGP and CHH do not seem to share a common genetic architecture. 38
Meanwhile, clinicians often fall back on the classic dogma of expectant management, that is, allowing adolescents with CDGP to undergo spontaneous initiation of puberty, as a means to identify those with CHH who do not. 39 As a consequence, the diagnosis of CHH is typically made in late adolescence or early adulthood, 40 leading to median 5 year delay in the initiation of pubertal induction. Moreover, around one-third of CHH patients have arrested partial puberty at presentation (testes ⩾ 4 ml, or breast stage 2–3) and may thus be wrongly reassured that ‘as puberty has begun, it will necessarily proceed to completion’.
Hence the crucial importance of identifying the presence of ‘red flags’ in a young man with pubertal delay, in particular cryptorchidism, micropenis, deafness, clefting or anosmia, would be strongly indicative of CHH. 41
Adults
Infertility and sexual dysfunction, including poor libido, erectile dysfunction and loss of spontaneous erections, are important manifestations of hypogonadism in adulthood. Nonspecific symptoms such as fatigue, reduced energy, low mood, inability to focus and sleep disorder can also be reported by hypogonadal men. Uncommonly, CHH may be uncovered in under-virialized adult men. 42 Such delayed presentation beyond teenage years could be due to reluctance to engage with healthcare systems, or failure to be supported with appropriate pubertal induction by physicians they encountered earlier in life, but is underpinned by minimal exposure to reproductive endocrinology in both undergraduate and postgraduate medical training.
To confirm the diagnosis of hypogonadism unequivocally and consistently, low serum testosterone must be demonstrated. 43 As circulating testosterone levels are significantly influenced by diurnal rhythm, food intake and changes in health status, venipuncture should be carefully scheduled to avoid false positive results. This is especially crucial in those with less specific clinical features of androgen deficiency to avoid misdiagnosis. Hence, it is recommended that blood specimens be obtained on two separate mornings (8–10 am) after an overnight fast, and testing should be deferred in the event of acute illness, altered sleep–wake cycle (e.g. shift work), or short-term use of medications (e.g. glucocorticoids, opioids) that can suppress testosterone concentrations. In addition, measurement of sex hormone-binding globulin (SHBG) to determine free testosterone concentrations by validated formula is helpful in men with conditions that can alter SHBG concentrations, such obesity and diabetes mellitus, or when initial total testosterone concentrations are equivocal. In the subsequent workup of HH, magnetic resonance imaging (MRI) of the sella turcica should be considered in men with broader pituitary hormone deficiencies, persistent or marked hyperprolactinaemia, profoundly low serum total testosterone (<5.2 nmol/l), 44 or symptoms of tumour mass effect (e.g. visual field defect, new-onset headache). If fertility is a concern, a minimum of two semen analyses should be performed in accordance to international standards established by the World Health Organization (WHO), 45 unless of course baseline TV is so low (<5 ml) as to make spermatogenesis extremely unlikely.
Treatment strategies
Infants
Neonates affected by cryptorchidism or micropenis must receive timely treatment to optimize genital development. Importantly, cryptorchidism has a far-reaching negative impact on future fertility potential, and delay in orchidopexy has been associated with further compromise in spermatogonia. 46 Therefore, current recommendations advocate surgical correction of undescended testis by about 1 year of age. 47 As for micropenis, the treatment usually entails a short course of low-dose testosterone delivered either by intramuscular injection or topical application to induce penile growth. A commonly described regimen is the intramuscular administration of four doses of 25 mg of testosterone cypionate or enanthate once every 3 weeks for 3 months. 48 Because the treatment is restricted to a brief duration, no significant concern exists with regards to virilization.
However, there are limitations to these treatments. Small testes render orchidopexy technically challenging with excess risk of testicular trauma and tissue loss, 49 which would further exacerbate the pre-existing testicular dysfunction. In addition, while exogenous testosterone effectively stimulates penile growth, it has absolutely no effect on testicular development. Recognizing the critical role of minipuberty, the feasibility and benefits of gonadotrophin treatment in recreating the hormonal milieu in affected male infants have been studied.
In the first published report of a boy with CHH and micropenis who received short-term recombinant human LH and FSH from age 7.9 to 13.7 months, the penile length successfully increased by 50% and the TV nearly tripled by the end of treatment. 50 In another report of two male infants (one case each of CPHD and CHH), 6 months of gonadotrophin combination therapy via subcutaneous pump infusion initiated at age 8 weeks and 20 weeks, respectively, led to a four-fold increase in both penile length and TV. 51 More recently, 3–6 months of continuous subcutaneous infusion of recombinant human gonadotrophins in five male infants (four CHH and one CPHD) produced a several-fold increase in serum IB concentrations, TV, and testosterone production. 52
More intriguingly, there is emerging evidence that spontaneous testicular descent could be successfully induced by gonadotrophin treatment, and thus obviate the need for surgery. In a study of eight infants with maldescended testes due to underlying HH (five CHH and three CPHD), gonadotrophin infusion induced complete testicular descent in six boys and partial descent in two boys, such that only one had to undergo orchidopexy eventually because of re-ascent of both testes 11 months later. 53 In another study, combined recombinant FSH and LH in the first 6 months of life successfully induced spontaneous testicular descent in two of four CHH/CPHD boys with bilateral cryptorchidism. 52 However, the evidence base is diluted by studies wherein gonadotropin treatment was used on ‘all-comers’ with cryptorchidism, rather than in boys with proven or suspected CHH.
Even if gonadotrophin treatment turns out to be unsuccessful in inducing spontaneous testicular descent, it would still be hugely beneficial in facilitating orchidopexy in gonadotropin-deficient boys by enlarging the TV presurgically.
Therefore, short-term neonatal gonadotrophin treatment appears to be promising in replicating the effects of minipuberty, by correcting micropenis, promoting testicular growth, and inducing descent of malpositioned testes, with the prospect of augmenting future sexual and reproductive function. 49
Adolescents
Pubertal delay is associated with considerable psychosocial impairment, including low self-esteem, social withdrawal, poor school performance and even higher rates of substance use disorder.33,54,55 This is especially problematic for CHH patients, as most only start to receive meaningful treatment in late adolescence at a mean age of 18–19 years. 56 The emotionally distressing experience of being stuck in a prepubertal, under-masculinized state while their peers progress through puberty can be psychologically scarring. 14 Furthermore, the embarrassment of immature external genitalia can produce adverse lasting impact on their sexuality and ability to form intimate relationship, especially for those with history of cryptorchidism or microphallus.56,57 Consequently, a high prevalence of men with CHH suffer from chronic depressive symptoms as adults, often requiring the use of antidepressants.56,57
Therefore, the tendency to indiscriminately feed teenagers with pubertal delay and ‘red flags’ for CHH into ‘delayed puberty of unknown cause’ management pathways 39 is inappropriate and potentially damaging. Indeed, a 12–13 year-old with absent puberty and ‘red flag’ CHH features, for example, anosmia, or history of cryptorchidism, should obviously be diagnosed as ‘CHH until-proven otherwise’ and receive immediate sex hormone therapy to initiate puberty.
If there are no suspicious features of permanent hypogonadism, and a presumptive diagnosis of CDGP is made following initial evaluation, treatment with the aim of accelerating growth or inducing secondary sexual characteristics should be discussed, especially for those who are facing psychosocial difficulties. Treatment is commonly initiated at intramuscular injection of 50 mg testosterone ester each month for 3–6 months with close follow up, 41 although this dosimetry should not be extrapolated for use in older apubertal men. 42 However, if there is persistent absence of endogenous gonadotrophin-dependent puberty (lack of testicular enlargement) on serial follow up, MRI brain with coronal T2 sequences through the olfactory bulbs would be indicated. In addition to excluding hypothalamo-pituitary pathologies, the radiological finding of absent or hypoplastic olfactory bulbs is pathognomonic for Kallmann syndrome.
For adolescents with CHH (or other cause of permanent HH), the testosterone dose is gradually increased over a course of 3 years until full adult replacement levels. Such a pubertal induction strategy seeks to replicate the typical tempo of puberty so that the appearance of secondary sexual characteristics would be age appropriate and in tandem with the degree of psychosexual development. 41 This is also necessary to ensure the optimization of statural growth by promoting a growth spurt and preventing premature fusion of epiphyseal plates. However, for older men in whom there are no concerns about final height, a faster tempo of pubertal induction is appropriate. 42
However, exogenous testosterone therapy does not induce testicular growth nor promote spermatogenesis. In clinical practice, teenage and young adult patients are often troubled by negative body image and poor self-esteem due to the appearance of small testes, despite having achieved full virilization in all other aspects. Notably, in a recent study on young men with HH, gonadotrophin treatment was found to have a greater positive impact on the health-related quality of life compared with testosterone replacement, particularly if spermatogenesis is successfully demonstrated on semen analysis, 58 which conceivably relates to an enhanced sense of masculinity. Moreover, early induction of spermatogenesis has the advantage of reducing the time required for appearance of sperm and the need for prolonged cycles of gonadotrophin treatment in adult life. As such, gonadotrophin treatment plays an essential role in the management of patients with prepubertal-onset HH and should be carefully planned for in order to optimize testicular maturation and fertility potential.
One important aspect to consider is the sequence of gonadotrophins initiation. In normal pubertal development, Sertoli cells and seminiferous tubules initially expand under the stimulation of FSH, and as intra-testicular testosterone increases with the progression of puberty, they stop proliferating and undergo terminal differentiation. Indeed, FSH monotherapy in children and adolescents with prepubertal-onset HH has been proven to be effective in promoting testicular growth and increase in circulating IB concentrations.59,60 Importantly, in a small subgroup of adolescents with CHH, FSH-priming prior to combination with hCG successfully induced spermatogenesis in three out of four patients. 60 This sequential gonadotrophin therapy, FSH monotherapy followed by combined FSH and hCG, is emerging to be a promising fertility-induction strategy. Crucially, as hCG monotherapy would cause premature differentiation of the diminished pool of Sertoli and germ cells, thereby abolishing any chance of proliferation, it should be avoided in those with immature testes.
As patients approach late adolescence/adulthood, close collaboration between paediatric and adult endocrinologists is necessary to ensure smooth transition of care, which is unfortunately an often neglected aspect in the care of young patients with chronic diseases. 57
Adults
Fertility can be restored in men with HH by sperm-induction protocols that replicate endocrine regulation of spermatogenesis using pulsatile GnRH therapy or exogenous gonadotrophins. However, while treatment successfully stimulates sperm production in majority of patients, largely comprises of adult (postpubertal)-onset HH, a significant proportion of men with congenital forms of HH have very poor response. These men have lifelong profound deficiencies in LH and FSH secretion which have severely compromised their spermatogenic potential. It is important to identify these individuals so that targeted treatment approach can be undertaken to improve outcomes.
Conventional sperm-induction regimens
Although pulsatile GnRH treatment is biologically and technically feasible to induce fertility in men with CHH (GnRH deficiency), it is rarely used outside of research settings because of limited availability of drug, infusion devices and expertise to deliver such a therapy. Moreover, it is ineffective in men with HH due to pituitary disease, hence further restricting its applicability. Therefore, exogenous gonadotrophins, which have importantly demonstrated comparable efficacy to that of pulsatile GnRH treatment,61,62 are favoured in clinical practices.
(i) hCG monotherapy ± adjuvant FSH (hCG ± FSH)
This regimen is only suitable for men at the mildest end of the disease spectrum, for example, men with adult-onset HH who have undergone normal pubertal and gonadal development, especially if they have fathered children before. In these men, treatment with only hCG is usually sufficient to achieve optimal sperm counts by about 6 months. Thereafter, if the sperm count remains unsatisfactory despite good adherence and appropriate dosing, FSH can be added to the regimen. However, a trial of hCG monotherapy is futile in men with prepubertal testes (TV <4 ml) or a history of cryptorchidism, as the failure rate is exceedingly high, even with prolonged therapy up to 10 years.63–65
As a long-acting LH-analogue, hCG is typically initiated at 3000–5000 IU per week divided into 2–3 subcutaneous injections. Common side effects to monitor for include erythrocytosis and gynaecomastia. Hence, to ensure well-tolerated and optimal dosing, clinicians should monitor trough levels of serum testosterone, haematocrit and oestradiol every 4–6 weeks to guide dose titration until a steady state has been achieved. Of note, as testes enlarge during the course of treatment, normal serum testosterone may sometimes be attainable at lower doses. Similar dosing principles would apply for newer recombinant hCG (choriogonadotrophin alfa), which comes in the form of injection prefilled pen. In resource-limited settings, careful storage of the remaining hCG in the refrigerator in between injections is considered safe.
(ii) Combined gonadotrophin treatment (hCG + FSH)
Depending on the severity of FSH and LH deficiencies, men with CHH (or CPHD) could have different prognosis for fertility outcomes. Those with partial gonadotrophin deficiencies generally have milder phenotype. They may enter puberty spontaneously, accompanied by a degree of testicular maturation, only to experience failure to progress later. 66 On the other hand, two-thirds of men with CHH have profound GnRH deficiency, characterized by prepubertal TV (<4 ml), low serum IB concentration (<60 pg/ml) or a history of cryptorchidism, which likewise are negative predictors of treatment response.27,67
For CHH patients with evidence of prior gonadal development (TV ⩾4 ml) and no history of cryptorchidism, combined gonadotrophin therapy (hCG + FSH) at inception is recommended to optimize testicular growth and spermatogenesis. However, in a subset of patients with near-adult size testes, hCG monotherapy could achieve success, with the addition of FSH after 6 months if response is suboptimal. Commonly available FSH preparations, highly purified urinary gonadotrophins (hMGs) and recombinant FSH, are self-administered by subcutaneous injections. The starting dose of FSH is 75 IU 3 times per week, with dose adjusted (up to 300 IU 3 times per week) as necessary, to achieve serum FSH concentration in the physiologic range of 4–8 IU/l. 68 If a new long-acting FSH analogue is chosen, injection is given every 10–14 days.68,69
As already stated, hCG monotherapy has no place in the management of men with prepubertal testes (TV < 4 ml) or a history of cryptorchidism, as the failure rate is exceedingly high, even with prolonged therapy63–65 and non-responders to hCG/FSH combination therapy after 3–6 months of hCG pretreatment (a protocol originally devised for FSH product licensing studies in men and not grounded in developmental physiology) are over-represented by patients with similar characteristics. 70 Therefore, spermatogenesis induction with combined gonadotrophin treatment from the outset is considered the standard of care for this group of patients. However, it is disappointing that up to one-third of men remains azoospermic on combined gonadotrophin therapy (or the equivalent pulsatile GnRH therapy). 62 Of the responders, a significantly longer period of treatment compared with men with larger baseline TV would be required before sperms are observed in ejaculate. 27 In addition, sperm counts typically plateau well below that of WHO reference value (⩾15 million/ml),45,27 although that does not necessarily preclude the possibility of natural conception.67,71,72
Sequential gonadotrophin treatment (FSH FSH + hCG)
Men with complete CHH have markedly diminished spermatogenic capacity due to the absence of FSH-stimulated Sertoli and germ cell proliferation, which would explain the dismal response to conventional gonadotrophin treatment. As such, it would be desirable to maximize the growth of Sertoli cells and seminiferous tubules first by administering FSH therapy only (FSH-priming), before the subsequent introduction of hCG therapy, so as to prevent premature differentiation of Sertoli cells under the influence of LH-induced intra-testicular testosterone.
The potential advantage of such unopposed FSH treatment in adult was demonstrated in a randomized, open-label trial of 13 treatment-naïve adult CHH men with prepubertal testes (TV < 4 ml). 73 In this study, seven men were randomized to recombinant FSH pretreatment for 4 months before embarking on a 24-month GnRH treatment protocol. During the FSH-only phase, TV doubled and serum IB levels rose to adult levels, and all patients in this arm subsequently developed sperm in ejaculate on GnRH therapy, compared with four of six men in the 24-month GnRH-only arm. Encouragingly, trends towards larger TV, higher maximal sperm counts and shorter time to first appearance of sperm in the ejaculate in the FSH-pretreated group were also observed.
Although larger multicentre studies are needed to confirm these findings, FSH-priming appears to be a logical and promising approach to improving fertility in men with a severe form of CHH, by providing a critical window for maximal expansion of immature Sertoli and germ cell mass. As serum IB was observed to plateau following 2 months of unopposed FSH treatment in the aforementioned study, 73 it can be safely inferred that treatment between 2–4 months would suffice, before introducing hCG to induce maturation and initiate the process of spermatogenesis. Once TV reaches around 8 ml, seminal fluid analysis can be performed every 2–3 months to monitor for sperm production.
Genetic counselling and screening of offspring
Genetic counselling is essential for patients with CHH who are seeking fertility because of the risk of passing on the disease to their offspring. 12 However, unless classical Mendelian inheritance mode of transmission (autosomal recessive, autosomal dominant or X-linked transmission) is certain, counselling is often difficult due to the complex genetic architecture (oligogenicity) in many cases. An estimated 5–10% transmission risk can be advised in such scenarios. 74 It is also good practice to have patients engaged with their providers/genetic specialists in ongoing discussion, as the genetic landscape is constantly evolving in the current era of next-generation sequencing. Furthermore, as screening of offspring for minipuberty is extremely helpful in facilitating early diagnosis, it should be offered to prospective parents during the counselling process.
Artificial reproductive technology and sperm cryopreservation
Spermatogenesis induction may take between 6 and 24 months to achieve maximal effect,62,72,75 and natural conception may not occur until 2–3 years of gonadotrophin therapy, 72 with significantly lower chance of success in men with poor prognostic factors. Therefore, given the potential length of treatment process, it would be necessary to factor in the urgency to conceive, particularly in relation to the age and health of the partner. Early involvement of a fertility specialist is thus important to facilitate concurrent evaluation for any female subfertility issue, as well as timely access to artificial reproductive technologies (ARTs) if needed, including in vitro fertilization, intracytoplasmic sperm injection (ICSI) and microdissection with testicular sperm extraction (micro-TESE). Compared with other surgical sperm retrieval techniques, micro-TESE has proven superiority in patients with nonobstructive azoospermia (e.g. Klinefelter syndrome) because of the direct magnified visualization afforded during the harvesting process, which would similarly be advantageous to men with HH should gonadotrophin treatment fail to achieve sperm in ejaculate. 76
If conception is successful, gonadotrophin treatment into the second trimester of pregnancy is recommended in order to maintain spermatogenic capability in the unfortunate event of miscarriage. Treatment with hCG alone may also be continued if the couple desires another child in the near future. 77 Otherwise, if decision is made to switch to testosterone replacement, sperm cryopreservation (banking) prior to stopping gonadotrophin therapy should be considered, as future fertility with repeat gonadotrophin treatment, though associated with quicker initiation of spermatogenesis,67,72 is not assured.
Further considerations
Long-term management of classical hypogonadism
Approximately 10–20% of men with CHH experience reversal of hypogonadism, albeit with risk of relapse. 78 Otherwise, the vast majority of men with organic HH would require lifelong physiological testosterone replacement to avoid androgen deficiency-related end-organ deficits, including anaemia, osteoporosis and muscle wasting. Among the formulations available, long-acting testosterone undecanoate depot injection and transdermal applications are preferred because of their favourable pharmacokinetics in providing stable circulating testosterone concentrations. Once appropriate dosing has been established, annual monitoring of testosterone, haematocrit/haemoglobin for testosterone-induced erythrocytosis, as well as prostate-specific antigen (PSA) in men aged ⩾55 years (40 years if high risk) is recommended. 43 In addition, baseline bone mineral density (BMD) measurements is useful to identify patients with low bone mass; testosterone replacement in hypogonadal men has been shown to be effective in improving BMD, 79 and thus antiresorptive agents should be deferred until later life, unless the patient has already sustained a fragility fracture.
Alternative treatment for functional HH: aromatase inhibitors and selective oestrogen receptor modulators
In recent years, there is increasing interest in the use of aromatase inhibitors (AIs) and selective oestrogen receptor modulators (SERMs) as alternative modalities in the treatment of male hypogonadism and subfertility. 80 It is crucial to recognize that, in order for these agents to work, hypothalamic–pituitary function must be intact because these act by decreasing oestradiol-mediated negative feedback inhibition. Therefore, AIs/SERMs are only potentially viable in patients with functional, but not organic, causes of HH. Some common scenarios include severe obesity, chronic opiate use, and withdrawal from AASs. But in the absence of robust data on the safety and efficacy of these agents, measures to address the underlying cause and avoidance of testosterone treatment are preferred instead. However, if aetiological factors cannot be resolved satisfactorily, or recovery of spermatogenesis does not occur within a reasonable timeframe, an off-label trial of AIs/SERMs for a limited course (e.g. clomiphene 25 mg, three times per week to 50 mg daily, anastrozole 1 mg, three times per week) 80 might be considered for selected patients before embarking on definitive fertility-induction treatment with gonadotrophins.
Conclusion
While infertility due to male HH is generally considered to be amenable to hormonal therapy, the clinical response depends principally on the onset and severity of the neuroendocrine defect. Men with congenital forms of HH typically have much less favourable prognosis because of severely compromised testicular development beginning in foetal life. Strategies to identify these patients early in childhood, as well as instituting timely pubertal induction in adolescence, can significantly improve long-term sexual and reproductive function. Furthermore, a fertility-induction protocol that seeks to maximize Sertoli and germ cell proliferation (FSH-priming) holds promise in enhancing spermatogenesis for these individuals. Couples would also benefit from multidisciplinary care and access to ARTs when necessary. Long-term physiological testosterone replacement is necessary for men with organic/permanent HH, whereas condition-specific treatment for those with functional HH can effectively restore endogenous HPT axis function.
Footnotes
Acknowledgements
Both authors contributed equally to the writing, editing and approval of this commissioned manuscript.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not for-profit sectors.
Ethical approval
Ethical approval was not required for this review.
Conflict of interest statement
The authors declare that there is no conflict of interest.