
Abstract
Esophageal organoids are self-assembled 3D microtissues derived from pluripotent or adult stem cells. They accurately recapitulate the cellular composition, tissue arrangement, and functional properties of the native esophagus. Emerging as powerful tools in disease research, they overcome key limitations of 2D cultures and animal models, such as poor microenvironmental simulation, species differences, and ethical concerns. Based on literature published in the past decades, this review summarizes recent advances in esophageal organoid construction, characterization, and applications in developmental biology, disease modeling, drug screening, and regenerative medicine. The current challenges, such as standardizing culture procedures, genetic instability, and microenvironmental complexity, are critically analyzed. We also highlight innovations such as microfluidic integration and high-throughput automation that improve model fidelity (accuracy and reliability) and support clinical translation. Future interdisciplinary efforts are expected to expand the role of esophageal organoids in precision medicine, enabling personalized diagnosis and therapy for esophageal disorders.
Introduction
The esophagus is a vital part of the digestive system. It transports food and acts as a barrier against harmful external factors. Its unique multilayered structure and specialized cellular microenvironments are essential for maintaining tissue homeostasis and responding to diverse pathological insults. 1 Clinically, esophageal diseases comprise a wide range of conditions, including congenital anomalies (e.g. esophageal atresia and tracheoesophageal fistula); chronic inflammatory disorders (e.g. eosinophilic esophagitis); premalignant lesions such as Barrett’s esophagus; and malignant tumors, primarily esophageal squamous cell carcinoma and esophageal adenocarcinoma. 2 These conditions are often characterized by complex pathogenic mechanisms and variable clinical presentations, which pose significant challenges to diagnosis and treatment. For example, although diagnostic and therapeutic approaches for esophageal cancer have improved, the overall prognosis remains poor. 3 Barrett’s esophagus, a premalignant precursor of adenocarcinoma, remains poorly understood regarding its origin and progression. 4 Similarly, congenital anomalies, even after surgical repair, often lead to postoperative complications that greatly diminish quality of life. 5 Collectively, these disorders highlight the urgent need for advanced experimental model systems to clarify disease mechanisms and to support the development of precision therapies.
Conventional two-dimensional cell culture systems are commonly used due to their simplicity. However, they inadequately mimic the esophagus’s complex three-dimensional structure and diverse cell populations. Moreover, they disregard the essential influence of the tissue microenvironment. 6 Animal models can partially reproduce the development of esophageal diseases; however, differences in physiology between species and ethical constraints restrict their translational applicability. 7 These models fail to accurately represent the cellular characteristics and disease complexity of the human esophagus, which hinders progress in fundamental and clinical studies.
In recent years, organoid technology derived from pluripotent and adult stem cells has led to significant advances in esophageal research. Organoids are three-dimensional microstructures formed by self-organization of tissue-specific stem cells or reprogramed pluripotent stem cells. They faithfully reproduce the cellular diversity, spatial arrangement, and functional properties of native tissue. 8 Compared to traditional models, organoids accurately replicate the multilayered squamous epithelium and proliferation-differentiation gradient of the esophagus, 9 and facilitate dynamic interactions between diverse cell populations and the microenvironment, 9 thereby greatly increasing the physiological relevance of disease research. Notably, organoids exhibit strong proliferative capacity and compatibility with high-throughput screening, offering a unique platform for disease modeling, drug discovery, and individualized therapy assessment. 10
Organoid technology is a novel method for modeling human organs and is gaining prominence in the field of precision medicine. Esophageal organoids, with their remarkable tissue-specific and functional characteristics, serve as robust platforms for investigating esophageal development, disease modeling, and assessing therapeutic compounds.9,11 By preserving patient-specific genetic and epigenetic profiles, esophageal organoids provide a reliable basis for elucidating genetic variations and disease mechanisms, thereby promoting clinical translation.12,13
This review systematically discusses methodologies for constructing and identifying esophageal organoids. It also highlights progress in their application for modeling tissue development and regenerative medicine. Furthermore, it examines novel applications of organoid technology in disease modeling, mechanistic studies, drug screening, and personalized therapeutic approaches for esophageal disorders. In addition, this review critically evaluates current technological challenges and future prospects. This work aims to provide comprehensive theoretical and practical guidance for investigators, to promote the clinical translation of esophageal organoids, and to facilitate advancements in the treatment of esophageal diseases (Figure 1).

Overview of esophageal organoid construction, applications, current challenges, and future prospects. This schematic summarizes the key elements involved in establishing and using human esophageal organoids. Construction and validation include the selection of cell sources and culture conditions followed by validation using morphology, marker expression, and functional assays. Major applications include studies of esophageal development and signaling pathways, disease modeling, drug screening, and regenerative medicine. Current challenges encompass standardization deficiency, genomic instability, inadequate microenvironment recapitulation, and dynamic functional gaps, which together contribute to barriers in clinical translation from bench to bedside. Future prospects include the development of esophageal organoid biobanks, organoid intelligence, full-thickness and complex models, immune co-culture systems, esophagus-on-a-chip platforms, and multi-organ integrated systems.
Construction and validation of esophageal organoids
The efficacy of esophageal organoid generation depends on selecting optimal cell sources (ranging from biopsies to pluripotent precursors) and tailoring the three-dimensional environment to mimic the native esophageal niche. To ensure model fidelity, it is essential to implement a comprehensive validation framework, encompassing analyses of morphological architecture, esophagus-specific molecular markers, and physiological-related functions verification. This section reviews the construction strategies for esophageal organoids derived from both pluripotent stem cells and adult stem cells, and details the methodologies for structural and functional characterization, thereby providing essential theoretical and methodological foundations for future applications.
Construction of esophageal organoid models
Based on cellular origin, esophageal organoids are primarily categorized into two types. The first is includes pluripotent stem cell (iPSC)-derived organoids, which are induced to differentiate into the esophageal lineage by embryonic developmental cues. 14 The second is adult stem cell (ASC)-derived organoids, which rely on tissue-specific niche factors to sustain their self-renewal and differentiation capacities. 15
Esophageal organoids derived from pluripotent stem cells
Pluripotent stem cells (PSCs) include embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs). ESCs are isolated from the inner cell mass of the blastocyst, while iPSCs are produced through somatic cell reprograming.16,17 Specifically, both types of cells have the ability to undergo unlimited self-renewal and differentiate into multiple lineages. Since Takahashi’s 2006 study showed that somatic cells could be reprogramed into induced pluripotent stem cells (iPSCs) by transducing specific transcription factors, including OCT4, SOX2, KLF4, and c-MYC, these cells have emerged as the main cell source in the field of organoid and regenerative medicine. They bypass embryo sourcing and related ethical issues,17,18 which makes them highly advantageous. Moreover, iPSCs can be generated from patients, minimizing the risk of immune rejection and making them highly attractive for disease modeling and tissue engineering applications.17,19 Pluripotency in iPSC systems is sustained through the coordinated regulation of key transcription factors, such as OCT4, SOX2, and NANOG. These factors interact in a complex manner to maintain self-renewal in the undifferentiated state and to enable responsiveness to differentiation signals. 17 When cultured under suitable conditions, iPSCs remain undifferentiated and continuously proliferative. Upon stimulation by defined induction signals, they activate differentiation pathways and sequentially form various cell lineages, thereby establishing the basis for the in vitro generation of esophageal organoids.
The in vitro generation of esophageal organoids from iPSCs is a highly regulated and complex process. It predominantly utilizes differentiation protocols based on embryonic development 14 and biomimetic strategies. 20 By mimicking key signaling pathways 14 —such as Wnt activation, BMP inhibition, and retinoic acid (RA) action—as well as molecular events, 20 including foregut patterning and the formation of the basal-differentiating layer during embryonic esophageal development, precise differentiation of iPSCs into esophageal tissues and efficient organoid construction can be achieved. Notably, the esophagus originates from the definitive endoderm, and its development essentially belongs to foregut morphogenesis. 21
The generation of esophageal organoids follows a defined, multi-stage protocol. This process typically begins with the maintenance of undifferentiated induced pluripotent stem cells (iPSCs), followed by their sequential differentiation into definitive endoderm (DE), anterior foregut endoderm (AFE), dorsal anterior foregut endoderm (dAFE). Finally, these progenitor cells are differentiated into three-dimensional organoids (Figure 2). Each stage is driven by specific combinations of culture media and signaling factors. It is accompanied by characteristic changes in cell morphology and stage-specific molecular markers, which collectively coordinate the in vitro organogenesis program.

Schematic overview of generating human epithelial organoids from two different cell sources. (A)Organoids from iPSCs: Somatic cells are reprogramed to pluripotency to generate iPSCs, which are then differentiated into definitive endoderm (DE), anterior foregut endoderm (AFE), and dorsal anterior foregut endoderm (dAFE), followed by plating in 3D culture. (B) Organoids from ASCs: A tissue biopsy is minced and enzymatically digested. The resulting cell suspension is strained, counted, and plated in 3D culture to initiate organoid formation within days. (C) Differentiation timeline of iPSCs: Stepwise differentiation stages: DE induction (D0–D3), AFE differentiation (D3–D6), dorsal anterior foregut endoderm specification (D6–D13), and esophageal organoid maturation (D13+). Cytokines corresponding to each developmental stage are indicated.
First, activating endoderm-related signaling pathways is essential for maintaining and expanding undifferentiated iPSCs. 22 Activin A, a member of the TGF-β superfamily effectively initiates iPSCs differentiation into endoderm. 23 Furthermore, the Wnt signaling pathway is crucial for early iPSC differentiation into esophageal progenitor cells. Activators of the Wnt signaling pathway, like the GSK-3 inhibitor CHIR99021, promote iPSCs differentiation into endodermal cells, setting the stage for esophageal-specific differentiation. 24 Teo et al. demonstrated that adding Wnt or BMP signaling under serum-free conditions improves Activin induction efficiency and boosts the yield of endodermal cells. 23 Further studies, including those by Li et al., 25 have shown that TGF-β signaling is crucial for endoderm differentiation. Specifically, TGF-β signaling regulates mitochondrial biogenesis and metabolic reprograming of oxidative phosphorylation. By regulating mitochondrial biogenesis and metabolic reprograming, TGF-β signaling establishes a novel link between metabolism and cell fate determination, offering a theoretical foundation to improve protocols for endoderm-directed differentiation. After 3 days of treatment with Activin A, BMP4, and CHIR99021 from Day 0 to Day 3, the iPSCs gradually differentiate into well-defined, flat, sheet-like endodermal cells. This process has been accompanied by significant upregulation of the endoderm markers SOX17 and FOXA2, as well as downregulation of the pluripotency marker OCT4.23,26
Next, to efficiently obtain foregut endoderm expressing the esophageal marker SOX2 (Days 3–6), definitive endoderm cells were sequentially treated with several factors. These included FGF10 (a member of the fibroblast growth factor family), Noggin (a BMP signaling inhibitor), SB431542 (a TGF-β signaling inhibitor), Wnt-3a (a Wnt signaling agonist, applied from Days 3 to 5, and retinoic acid RA, added on Day 5). At this stage, the cells transitioned into a colony-like morphology and mainly consisted of slow-proliferating, flat, epithelial-like cells, with upregulated expression of the foregut marker SOX2. Studies have shown that foregut endoderm differentiation depends on the temporal regulation of signaling. 14 Specifically, Wnt signaling must be tightly restricted to a single day to prevent differentiation toward mid- and hindgut lineages. 14 RA signaling regulates foregut fate by activating HNF1β; short-term activation (1 day) promotes differentiation toward the anterior foregut, specifically the esophagus. 27 Rankin et al. critically guided the in vitro differentiation of esophageal organoids by temporally regulating the Wnt/BMP/RA signaling pathway. They first inhibited BMP to establish foregut identity, then activated RA to prepare the tissue for subsequent Wnt/BMP induction, ultimately specifying esophageal lineage. 28 Nyeng et al. proposed that FGF10 effectively promoted the differentiation of endodermal cells into esophageal progenitor cells. Meanwhile, it inhibited their differentiation into gastric or intestinal cell lineages. 29 Green et al. demonstrated that dual inhibition of TGF-β and BMP signaling helped endodermal cells acquire foregut endodermal characteristics. This process also ensures proper embryonic axial specification, that is, correct differentiation and spatial arrangement of endodermal cells along the embryo’s anterior–posterior and dorsal–ventral axes. This crucial step determines whether the organoid system can generate the correct foregut structure. 30 In addition, BMP inhibition maintains a low BMP signaling environment that promotes SOX2 expression, a key regulatory mechanism in foregut organogenesis, including esophageal development. Application of the BMP inhibitor Noggin simulates this low BMP signaling environment characteristic of foregut development. 31
Subsequently, anterior foregut endoderm cells are plated on Matrigel. From Days 6 to 9, continuous BMP inhibition and RA activation, combined with EGF and FGF10 supplementation, enhance the formation of dorsal anterior foregut endoderm. 14 At this stage, floating foregut spheroids, also described as small multicellular aggregates, emerge. This marks successful foregut specialization and represents a key milestone in acquiring esophagus-specific progenitor cells, laying the foundation for further three-dimensional structure. Differentiated spheroids increasingly expressed SOX2/P63 (basal stem cell markers) and KRT5/KRT14 (basal keratins). During treatment with EGF and FGF10 (Days 9–13), epithelial cell expansion is facilitated and organoid formation efficiency is enhanced. 20 Consequently, the spheroids display a well-organized multilayered structure with compact surface cell arrangement and occasional centrally located differentiated cells.14,20,32
Finally, spheroids were harvested and embedded in Matrigel, Cultrex or other extracellular matrices rich in laminin and collagen. This embedding initiated three-dimensional organoid culture. Within the three-dimensional matrix, spheroidal cells retained their prior signaling memory, rapidly polarized, and organized into a stratified epithelium. The organoids progressively expanded to sizes ranging from 200 to 500 μm. Some samples formed structures comprising 30–40 cell layers, with maximal maturity observed after 10–24 days of culture.14,20 For long-term esophageal organoid culture, routine passaging, density management, and continuous EGF supplementation are necessary. These measures allow maintenance of the culture in vitro for up to 2 months.
Esophageal organoids derived from adult stem cells
Adult stem cells (ASCs) are undifferentiated cells residing in mature tissues. They have the ability to self-renew and differentiate into specialized functional cell types. 33 Through in vitro three-dimensional (3D) culture, ASCs give rise to organoids that faithfully recapitulate the structural and functional characteristics of their source tissue. Compared with pluripotent stem cell (PSC)-derived organoids, ASC-derived organoids display distinct advantages, including stronger tissue specificity, greater genetic stability, a single-lineage composition, higher functional maturity, and faster establishment.34,35
However, current ASC-derived esophageal organoids, although exhibiting subtype heterogeneity within epithelial populations, are predominantly composed of epithelial cells and lack mesenchymal as well as other non-epithelial elements. 36
The esophageal epithelium consists of stratified squamous cells, including basal, intermediate, and terminally differentiated cells. Studies showed that adult stem cells typically reside within specialized microenvironments, and their functionality largely depends on these niches, known as the stem cell niche. 37 In the esophageal epithelium, the adult stem cell niche is located in the basal layer, where p63 and Sox2 act as core markers, and all epithelial cells originate from this region. 38 This cell population shows strong organoid-forming capacity and self-renewal, representing a typical stem cell group that serves as an optimal source for esophageal organoid construction. The stem cells exhibit robust self-renewal capacity. They generate transient amplifying (TA) progenitors within the basal layer through asymmetric division. 39 TA cells migrate upward and differentiate into various esophageal epithelial lineages. This process maintain epithelial homeostasis and facilitates tissue repair following injury.39,40
Successful cultivation of ASC-derived esophageal organoids requires carefully recreating the native tissue microenvironment to support stem cell proliferation and differentiation. This process involves three essential steps: isolating stem cells from esophageal tissue, optimizing the culture medium to reproduce in vivo signaling cues, and selecting appropriate three-dimensional scaffold materials. Collectively, these elements enable robust organoid growth and functional maturation 41 (Figure 2).
A critical initial step is the precise isolation of stem cells from esophageal tissue. DeWard et al. identified three functionally distinct subpopulations within the basal epithelium of the mouse esophagus using flow cytometry using the surface markers Itga6/Itgb4 and CD73. Among these, the Itgb4HighCD73+ population showed the highest organoid-forming efficiency and long-term self-renewal. Therefore, it represents a key candidate for esophageal organoid modeling. 40 Once isolated, these stem cells require optimized culture media to maintain stemness and form multilayered epithelial structures in vitro.
The composition of the culture medium plays a decisive role in normal organoid growth. Most media use DMEM/F12 as the basal formulation, which is supplemented with proliferation- and differentiation-associated factors. In most reported systems, Wnt/β-catenin signaling represents the core pathway maintaining stem cell identity. R-spondin acts as a potent Wnt signal amplifier and markedly enhances clonal formation and long-term expansion. In parallel, inhibition of bone morphogenetic protein (BMP) signaling is required to prevent premature differentiation. Noggin antagonizes BMP ligands and, together with Wnt signaling, maintains organoids in a proliferative and undifferentiated state. Additional components support epithelial stability and survival. The TGF-β pathway inhibitor A83-01 suppresses epithelial–mesenchymal transition (EMT) and stabilizes epithelial identity. Epidermal growth factor (EGF) provides mitogenic support. The ROCK inhibitor Y-27632 is typically applied transiently during seeding or early passaging to reduce apoptosis, while the B27 supplement supplies essential nutrients, including antioxidants, hormones, and lipids. Collectively, these components form the basic framework for normal esophageal organoid culture42 –44 (Table 1). Variations in medium composition across laboratories likely reflect differences in tissue sources and experimental objectives. More recently, simplified culture systems have been explored to define minimal signaling requirements. For example, protocols using keratinocyte serum-free medium (KSFM) as the basal medium support organoid formation through modulation of calcium ion concentrations combined with transient Y-27632 supplementation. 45 These approaches highlight the role of calcium signaling in regulating the epithelial proliferation–differentiation gradient but require further validation to ensure broader applicability and comparability.
Comparison of culture medium components for ASC-derived normal esophageal epithelium organoids.

Note. Antibiotics are auxiliary components commonly used in cell culture to prevent microbial contamination and do not exert direct biological effect on epithelial cells. “✓” indicates used, and “−” indicates not used or not reported.
The choice of scaffold is equally crucial, as it must effectively mimic the esophageal extracellular matrix (ECM). The rational selection of three-dimensional scaffolds is crucial in the construction of esophageal organoids. the core of which resides in mimicking the in vivo mechanical and biochemical extracellular matrix (ECM) microenvironment. Commonly used scaffolds include natural matrices like Matrigel and tunable synthetic hydrogels. Natural matrices like Matrigel, rich in ECM components such as laminin and type IV collagen, effectively support the growth of diverse gastrointestinal organoids; however, their compositional variability and lot-to-lot differences limit mechanistic studies and clinical translation. 46
To address these limitations, fully defined, mechanically tunable synthetic hydrogels—such as PEG-, PIC-, or hyaluronic acid (HA)-based systems—have been developed and demonstrated efficacy in epithelial and gastrointestinal organoids as viable alternatives or adjuncts to Matrigel.47,48 In esophageal adenocarcinoma (EAC), Cruz-Acuña et al. developed NorHA, a modular HA-based synthetic hydrogel with stiffness precisely tunable via crosslinking density, enabling independent incorporation of adhesion peptides and degradable sequences. This platform facilitates robust, long-term 3D culture of patient-derived EAC organoids (PDOs). In the context of chemically defined cues, ~1000 Pa tumor-associated matrix stiffness proved sufficient to independently promote EAC organoid proliferation, morphological expansion, and activation of stemness-associated transcriptional programs, thus elucidating the pivotal role of matrix mechanics in EAC progression. 49 Scaffold attributes—including 3D network architecture, porosity, and plasticity, alongside ECM proteins, adhesion ligands, and growth factors—collectively dictate esophageal organoid morphogenesis, apico-basal polarity, and functional differentiation. Ideally, such scaffolds integrate tissue-specific ECM mimicry, dynamically tunable mechanics, and robust biocompatibility/translatability to recapitulate EAC microenvironments in vitro, advancing disease modeling and regenerative medicine.
Protocols for generating normal human esophageal squamous organoids typically proceed as follows: First, the esophageal mucosa and epithelial layer are harvested from fresh tissue. The muscularis is then removed. The basal epithelial layer is dissected, fragmented mechanically, and digested enzymatically (e.g. dispase and trypsin) to yield a single-cell suspension. These cells are resuspended in Matrigel and plated as domes in culture dishes. Cultures are maintained at 37°C in 5% CO2, with medium changes every 3 days. After 10–14 days, epithelial monolayers or early organoid structures emerge. These structures progressively differentiate into multilayered epithelia under the influence of supplemented induction factors. 9
These methods serve as the foundation for developing both normal and disease-specific esophageal organoids. Although the core culture principles remain similar, modifying the medium composition allows the system to model specific diseases or patient samples. Tumor organoids, for example, originate from cancer stem cells (CSCs) or self-renewing tumor cells that grow in 3D to form tumor-like structures. 50 This approach has enabled modeling of ESCC and EAC. It also allows the study of inflammatory and premalignant conditions, such as eosinophilic esophagitis and Barrett’s esophagus.9,51,52
Characterization of esophageal organoids
Esophageal organoids, as three-dimensional in vitro models, underwent comprehensive validation protocols. These included morphological assessment, molecular marker analysis, and functional assays to ensure their structural and functional reliability.
Morphological and histopathological identification
Hematoxylin and eosin staining was used to confirm that esophageal organoids mimic the morphological characteristics of the native tissue. Normal esophageal organoids must demonstrate the in vivo stratified squamous epithelial architecture, including basal, intermediate, and terminal differentiation layers with organized cellular alignment and distinct polarity; basal cells show cuboidal or columnar shapes, while superficial cells exhibit a flattened morphology. 53 Together, this architectural organization indicates epithelial stratification and reestablishes the physical basis of the esophageal barrier. In organoids modeling eosinophilic esophagitis and Barrett’s esophagus, disease-specific histopathological changes—including basal cell hyperplasia and columnar epithelial metaplasia—were required to be reproduced.9,52 Organoids derived from esophageal squamous cell carcinoma and adenocarcinoma were expected to exhibit corresponding neoplastic characteristics, such as nuclear atypia, keratin pearl formation, and polarity disruption.13,44
Organoids derived from disease models such as eosinophilic esophagitis (EoE) and Barrett’s esophagus (BE) are expected to recapitulate disease-specific histopathological alterations. Specifically, EoE organoids exhibit a marked thickening of the basal cell layer, disruption of the epithelial differentiation gradient, and thinning of the superficial keratinized layer. These changes collectively indicate an impairment of epithelial barrier function. 54 BE organoids consist of a single layer of columnar epithelial cells with mucus-secreting goblet cells and express markers typical of both pyloric gland and intestinal differentiation, thereby exhibiting the phenotype of intestinal metaplasia. 52 Patient-derived organoids (PDOs) from esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC) generally preserve morphological and pathological features consistent with their corresponding primary tumors. ESCC organoids typically consist of atypical squamous cells characterized by a high nuclear-to-cytoplasmic ratio, active proliferation, and aberrant p53 accumulation. These organoids exhibit pronounced nuclear atypia and poorly differentiated morphological features. They faithfully recapitulate tumor-like architectures in three-dimensional culture while exhibiting limited squamous differentiation. 42 In contrast, EAC organoids commonly display disorganized epithelial structures, formation of multiple luminal cavities, and loss of organized apical-basal polarity. This reflects disruption of cellular polarity, a hallmark of esophageal adenocarcinoma. 13
Molecular markers
In the study of esophageal organoids, molecular marker identification was predominantly performed to localize cell type–specific protein expression using immunohistochemistry and immunofluorescence. In normal esophageal organoids, basal stem cell identity was confirmed via markers p63, SOX2, and KRT14, while mid-layer and terminally differentiated cells expressed KRT10, KRT13, and involucrin, ensuring sequential differentiation. 55 In disease models, squamous cell carcinoma organoids expressed tumor-associated markers CK10, p63, and Ki67, 56 whereas adenocarcinoma organoids expressed glandular markers CK7, CK20, and CDX2, 13 underscoring subtype-specific molecular features.
Multi-omics analyses were employed to elucidate organoid gene expression profiles and regulatory mechanisms. Normal organoid gene expression closely matched native tissue, exhibiting high levels of squamous differentiation genes (KRT5, TP63) and barrier genes (CLDN1, OCLN), while excluding tracheal and gastric markers to maintain tissue specificity. 57 In pathological models, multi-omics approaches uncovered disease-associated gene dysregulation and regulatory network alterations, providing a molecular foundation for pathogenesis. 13 TP53 mutations are prevalent in ESCC organoids, and alterations in additional driver genes, such as CDKN2A, are likewise maintained. Whole-exome sequencing (WES) analyzes confirm that these key mutations remain stable during prolonged in vitro culture. 44 In EAC organoids, whole-genome sequencing (WGS) reveals that driver mutations in TP53, CDKN2A, and PIK3CA, together with structural variants such as chromothripsis, receptor tyrosine kinase (RTK) amplifications, and copy-number alterations, exhibit a high degree of concordance between organoids and the corresponding primary tumors. 13
Function verification
During functional validation, esophageal organoids were demonstrated to emulate essential barrier functions and response mechanisms to external stimuli under both physiological and pathological conditions. Transepithelial electrical resistance (TEER) measurements and FITC–dextran permeability assays were employed to evaluate the integrity of the esophageal epithelial barrier. 58 Tumor-derived organoids exhibited treatment-relevant functional characteristics as determined by drug sensitivity assays.44,59
Because the primary physiological role of the esophagus is to maintain a protective epithelial barrier, models for evaluating epithelial integrity are essential, while barrier functionality is often corroborated using an air–liquid interface (ALI) model. Quantitative techniques, such as transepithelial electrical resistance (TEER) measurement and fluorescent tracer permeability assays (macromolecular flux analysis), are commonly employed. Functional outcomes obtained from ALI models can be integrated with structural characteristics of organoid systems and are extensively utilized in studies of eosinophilic esophagitis (EoE) to characterize barrier dysfunction. Previous studies have demonstrated that epithelial cells derived from EoE patients show significantly lower TEER values and higher macromolecular flux after ALI culture compared to those from healthy donors, indicating intrinsic defects in epithelial barrier function. 58
The validation of pathological responses is achieved by reproducing common disease-associated conditions of the esophagus. Exposure of normal organoids to EoE-related cytokines, such as TNF-α and IL-13, can give rise to a basal cell hyperplasia (BCH)-like phenotype and suppress differentiation marker expression. 9 In gastroesophageal reflux disease (GERD) models, exposure of Barrett’s esophagus (BE) organoids to an acid–bile salt mixture promotes cell migration, reduces intercellular adhesion, and upregulates epithelial–mesenchymal plasticity (EMP)-related genes, thereby confirming their functional adaptability to reflux-induced stress. 52
The functionality of tumor organoids is primarily assessed through drug sensitivity assays to determine therapeutic responsiveness. Treatment of esophageal cancer (ESCC/EAC) organoids with agents such as cisplatin, 5-fluorouracil, or paclitaxel, followed by ATP-based viability measurement using the CellTiter-Glo assay or live/dead cell staining to determine the half-maximal inhibitory concentration (IC50), has been shown to effectively predict patients’ clinical responses to chemotherapy.42,60
Application of organoids
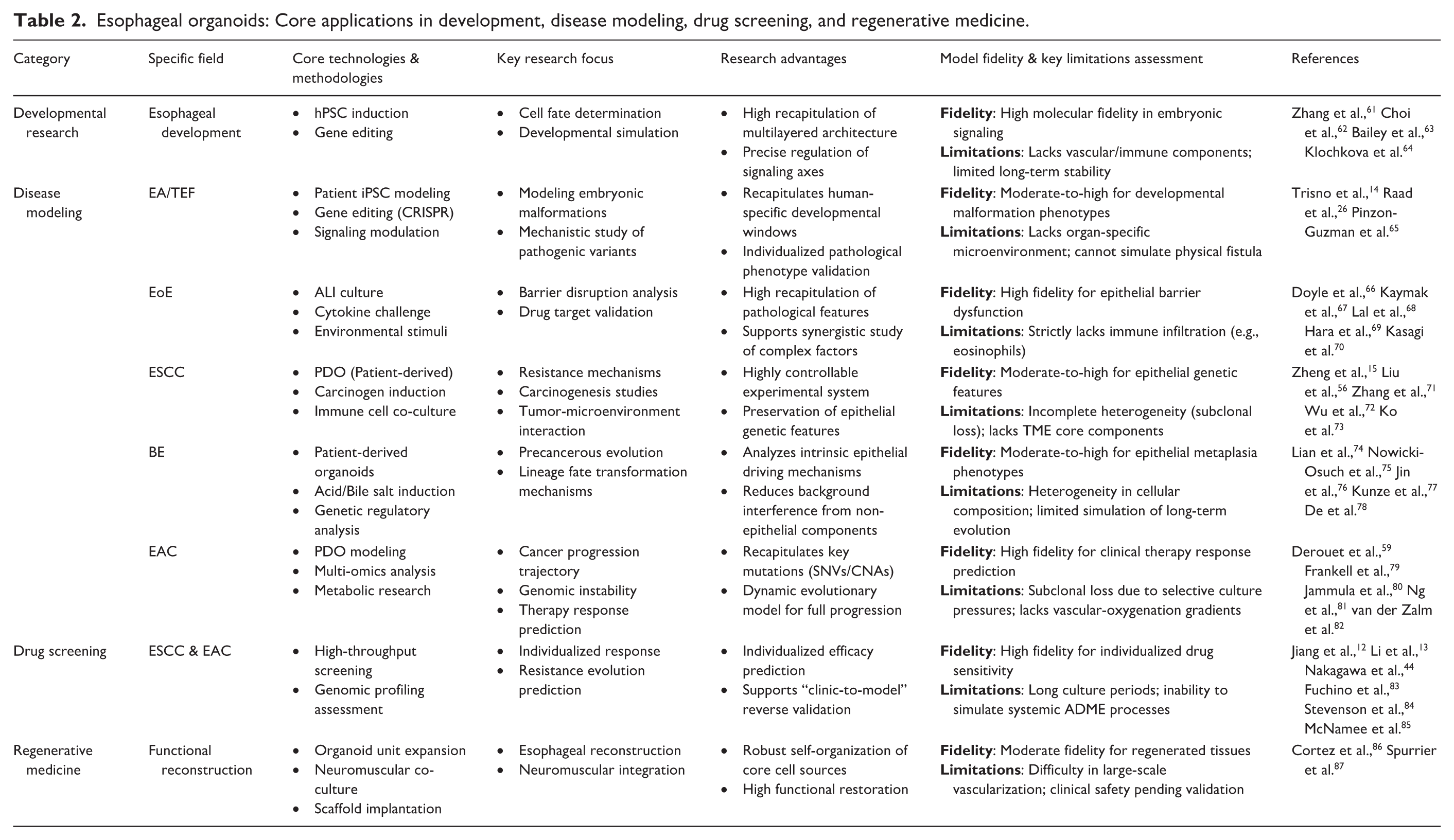
Esophageal organoids have shown broad application potential across multiple research fields. However, as simplified in vitro models, organoids have inherent limitations in simulating complex in vivo environments, which requires careful consideration when interpreting research findings and evaluating translational potential. This section systematically reviews the latest advances in the use of esophageal organoids in developmental biology, disease modeling, drug screening, and regenerative medicine (Table 2), with a particular emphasis on their critical role in advancing esophageal science and clinical research.
Esophageal organoids: Core applications in development, disease modeling, drug screening, and regenerative medicine.
Research on the mechanism of esophageal development
In vitro models of esophageal development have used organoids to reconstruct esophageal morphogenesis in three dimensions. The primary aim was to precisely recapitulate basal stem cell fate decisions and to simulate three-dimensional tissue architecture dynamically.58,61 Organoids derived from 3D cultures of basal epithelial stem cells faithfully reproduced stratified squamous epithelium and its proliferation–differentiation gradient, thus emulating key features of in vivo esophageal tissue.
Precise control of signaling pathways is essential for normal esophageal development and tissue homeostasis. Among these, the Hippo–YAP and Notch signaling pathways are pivotal regulators of cell fate determination and organ morphogenesis in the esophagus.
Regulatory role of the Hippo-YAP signaling pathway
In the esophageal epithelium, Hippo–YAP signaling controls the balance between basal stem cell proliferation and differentiation, thereby maintaining the structure and function of the stratified squamous epithelium. In mouse esophageal organoids, YAP1 is mainly located in stem cell nuclei and is essential for sustaining stem cell self-renewal. YAP1 inhibition reduces proliferative capacity and induces premature differentiation, demonstrating its role in preserving the undifferentiated state and regulating differentiation balance. 62 Moreover, human pluripotent stem cell-derived organoids and mouse genetic models confirmed that YAP deficiency significantly decreases basal epithelial cell proliferation, thins the epithelium, and impairs Krt13-positive suprabasal keratinocyte differentiation. YAP coordinates the basal-to-suprabasal transition by regulating Notch signaling, as shown by reduced NICD1 expression. Organoid experiments recapitulate this phenotype, validating YAP’s necessity in embryonic esophageal epithelial stratification and lineage specification. 63
Stage-dependent regulation by Notch signaling
During esophageal development, Notch signaling regulates basal progenitor differentiation into stratified squamous epithelium in a stage-dependent manner through the Jag2–RBPjk axis. Loss of Notch activity markedly impairs differentiation, lead to aberrant epithelial morphology, and highlights its critical role in esophageal formation and maintenance. Moreover, Notch signaling cooperates with BMP and other pathways to ensure proper differentiation and establishment of polarity in foregut endodermal cells. Notch activity is essential for maintaining esophageal epithelial homeostasis and barrier function by modulating basal stem cell proliferation and differentiation, preventing hyperplasia, and promoting tissue repair. Consequently, it is a key focus in esophageal disease pathogenesis and therapeutic research. 61
Novel autophagy mechanism in homeostasis maintenance
Klochkova et al. used mouse esophageal organoids, primary esophageal epithelial cells, and a tamoxifen-inducible, squamous epithelium–specific Atg7 conditional knockout mouse model. They exposed these models to 4NQO to systematically examine autophagy’s role in esophageal epithelial homeostasis. The study showed that ATG7-dependent autophagy maintains the esophageal proliferation–differentiation gradient by precisely regulating basal cell behavior, while ATG7 ablation leads to basal cell hyperproliferation and epithelial disorganization. Cyto-ID fluorescence–activated cell sorting revealed that basal cells with high autophagic vesicle levels (Cyto-IDHigh) exhibit restricted in vivo proliferation but enhanced self-renewal capacity; in organoid cultures, these cells displayed aberrant proliferation, including Ki67-positive cells beyond the basal layer. Mechanistically, Cyto-IDHigh cells exhibited activation of the autophagy pathway alongside cell cycle arrest, while their derived organoids were enriched for G2/M phase–associated gene expression. These findings indicate that autophagy functions not only as a cytoprotective mechanism but also as a microenvironment-responsive regulator of esophageal epithelial homeostasis. This offers novel insights into the pathogenesis of esophageal disease. 64
Disease modeling and mechanism research
Esophageal diseases encompass a variety of complex pathological conditions, including congenital anomalies, inflammatory disorders, and tumors. A deep understanding of their molecular mechanisms is essential for advancing precision diagnosis and therapy.
Current research mainly relies on models such as two-dimensional cell cultures, animal models (e.g. genetically engineered mice), and patient-derived xenografts (PDX). Although each system has certain advantages, they also exhibit notable limitations. Two-dimensional models fail to recapitulate the stratified architecture and tissue microenvironment of esophageal epithelium. 88 Animal models are limited by interspecies differences and cannot fully replicate the heterogeneity of human diseases. 89 PDX models are costly, low-throughput, and less suitable for detailed mechanistic dissection. 90
In this context, organoid models are capable of long-term expansion in three-dimensional cultures while retaining partial genetic and phenotypic features of the source tissue. They provide a novel platform to simulate disease-associated gene mutations, inflammatory stimuli, and epithelium-stroma interactions in a relatively controlled setting. Organoids are particularly valuable for elucidating intrinsic epithelial mechanisms and validating the functional effects of pathogenic mutations. However, for processes strongly dependent on immune microenvironment, neuromuscular regulation, or systemic factors, they still require complementary use with animal models or co-culture systems. This section will systematically review the modeling strategies and application progress of esophageal organoids in mechanistic studies, based on this defined role.
Congenital developmental abnormality disorders - Esophageal atresia/tracheoesophageal fistula
Esophageal atresia (EA) is the most prevalent congenital esophageal anomaly. It arises from abnormal foregut partitioning during embryogenesis and co-occurs with tracheoesophageal fistula (TEF) in 70–90% of cases. Surgical repair which includes esophageal anastomosis and fistula ligation, is the main treatment approach. Current survival rates exceed 90%, especially in patients without comorbidities who tend to have the best prognosis. Despite these advances, postoperative complications remain common and include esophageal dysmotility, anastomotic stricture, gastroesophageal reflux disease, and Barrett’s esophagus caused by reflux. These conditions collectively lead to dysphagia and significantly reduce long-term quality of life. 91 To tackle these clinical challenges, organoid technology has recently emerged as a pivotal tool .
The transcription factor SOX2 critically regulates esophageal embryogenesis and tissue homeostasis. SOX2 primarily maintains the stemness and phenotypic stability of foregut endodermal cells, which facilitates the formation and differentiation of the stratified squamous esophageal epithelium. Early in esophageal development, SOX2 was highly expressed in the dorsal foregut endoderm, inducing and sustaining esophageal progenitor lineage specification and preventing aberrant transdifferentiation toward respiratory epithelium. By modulating downstream targets, SOX2 activates esophageal epithelial differentiation markers and represses genes associated with tracheal or pulmonary phenotypes, thereby ensuring precise developmental patterning. Thus, spatial and temporal regulation of SOX2 expression is essential to delineate and stabilize the esophageal–respiratory boundary.92,93
Trisno et al. utilized human pluripotent stem cell-derived esophageal organoids combined with gene editing. They aimed to elucidate SOX2′s dual regulatory mechanisms in esophageal morphogenesis. Their findings demonstrated that SOX2 deficiency during dorsal–ventral foregut patterning impedes epithelial differentiation and induces a tracheal-like phenotype, which recapitulates the cellular identity dysregulation observed in EA/TEF. Moreover, sustained SOX2 deficiency during epithelial layer formation further compromised the development of multilayered squamous epithelium. This study SOX2 as a central determinant of esophageal cell fate. Furthermore, it provided a robust organoid platform for mechanistic investigations and therapeutic exploration in EA/TEF. 14
Building on these findings, Raad et al. generated patient-specific induced pluripotent stem cell (iPSC) models of EA/TEF. These models recapitulated key molecular events of human esophageal development in vitro. Patient-derived cells exhibited significant SOX2 downregulation during foregut differentiation, accompanied by ectopic NKX2.1 expression in mature organoids. This spatiotemporal SOX2 disruption led to an imbalance in esophageal–tracheal fate specification without impairing final differentiation and stratification of KRT4⁺/KRT13⁺ epithelium. Nanopore sequencing further revealed SOX2 dysfunction–associated dysregulation of multiple cooperating genes, including oxidative stress-related GSTM1 and vesicular transport gene RAB37. These findings implicate broader metabolic and organelle dysfunction in disease pathogenesis. 26
Pinzon-Guzman et al. investigated the role of the NOG/BMP signaling axis in esophageal development using human tissue specimens and organoid models. Esophageal organoid units (EOUs) derived from both mouse and human tissues, as well as tracheal organoid units (TOUs), demonstrated that NOG inhibits BMP4/SMAD1/5/9 signaling, sustaining squamous epithelial differentiation. Loss of NOG expression and aberrant BMP4 activation were observed in EA/TEF patient tissues, which caused foregut progenitor transdifferentiation toward a respiratory lineage. Treatment with NOG antagonist induced respiratory markers TFF3 and CC10, while suppressing squamous markers CK13 and CK4; recombinant NOG protein restored normal differentiation and reversed the aberrant phenotype. DNA methylation analysis suggested that epigenetic silencing of NOG may contribute to EA/TEF pathogenesis. These findings clarify the pivotal regulatory role of the NOG/BMP pathway in esophageal development and offer a foundation for developing targeted therapeutic strategies. 65
In summary, accumulating evidence suggests that the pathogenesis of esophageal atresia/tracheoesophageal fistula arises not from a single disrupted signaling pathway, but from early deviations in lineage specification of foregut progenitors that are progressively amplified during development, ultimately resulting in defective esophagotracheal separation. Esophageal organoid systems overcome key limitations of conventional two-dimensional cultures by recapitulating critical temporal stages of human foregut development and enabling modeling of lineage differentiation and stratified squamous epithelial formation in vitro.
Inflammatory diseases - Eosinophilic esophagitis
Eosinophilic esophagitis (EoE) is a chronic immune-mediated condition characterized by eosinophil infiltration of the esophageal mucosa, basal cell hyperplasia Its pathogenesis is multifactorial, involving various cytokines, signaling pathways, and epithelial dysfunction.94,95
In cytokine-driven epithelial injury, IL-33 functions as key alarmins contributing significantly to EoE development. Masuda et al. utilized IL-33 transgenic mice (EoE33) and organoid cultures to elucidate a key role of the IL-33/IL-13 axis in EoE pathogenesis. IL-33 overexpression caused basal zone hyperplasia, fibrosis, and esophageal motility impairment, with all pathological features strictly dependent on IL-13; IL-13 knockout eliminated these disease phenotypes. Organoid models confirmed that IL-33 alone triggers epithelial proliferation and differentiation abnormalities, whereas the full disease phenotype requires involvement of immune microenvironment. 96 Kaymak et al. employed patient-derived organoids to demonstrate that IL-20 subfamily cytokines decrease critical barrier proteins (FLG, FLG2), thereby impairing barrier integrity and facilitating EoE progression. MAPK/ERK signaling mediates these changes, and inhibition of this pathway mitigates experimental EoE. 67
Additional regulatory factors also influence the structure and function of Esophageal epithelium. Kasagi et al. 70 used organoid and co-culture models to show that fibroblast-derived TNF-α activates TGF-β/SMAD signaling, raises LOX expression in epithelial cells, and drives collagen cross-linking and tissue remodeling. Clinically, LOX levels correlate with EoE fibrosis severity, elucidating the “inflammation–fibrosis” pathway and suggesting potential targets for therapy.
Sasaki et al. 97 employed 3D organoids and single-cell sequencing. They found that LOX is upregulated in differentiated epithelial cells during IL-13 inflammation. This upregulation facilitates epithelial differentiation and barrier recovery through BMP pathway activation. LOX-overexpressing organoids showed reduced basal markers (TP63, SOX2, KRT14) and increased differentiation markers (IVL, FLG, LOR) expression, confirming the LOX/BMP axis in inflammatory epithelial homeostasis and revealing a new therapeutic target for EoE.
Environmental influences significantly affect esophageal epithelial barrier integrity in EoE. Doyle et al. showed that sodium dodecyl sulfate (SDS), damages the epithelial barrier and stimulates IL-33 release in air–liquid interface culture, organoid, models and mouse models, provoking EoE-like inflammation. This finding provided new insight into exogenous EoE triggers. Furthermore, microenvironmental hypoxia and its molecular regulation also influence the epithelial barrier function in EoE. 66 Markey et al. employed the EPC2-hTERT human esophageal organoid model to show that hypoxia-induced miR-155 impairs barrier function by downregulating the tight junction protein claudin-7. 98 Moreover, Lal et al. employed EPC2-hTERT organoid models to demonstrate that IFN-γ activates phosphorylation of STAT1, suppresses epithelial proliferation, disrupts barrier function, and triggers caspase-dependent apoptosis. CD8+ T cells expressing CD69, FOS, and ITGAE are the main local source of esophageal IFN-γ. Persistent infiltration of these cells may fuel chronic EoE inflammation and contribute to clinical heterogeneity. 68 These findings suggest that environmental factors and signaling molecules are central regulators of epithelial dysfunction in EoE.
Recent studies have advanced our understanding of epithelial progenitor cell involvement in EoE pathogenesis. Hara et al. identified a significant loss of regenerative CD73+CD104+ epithelial progenitors in the esophagi of EoE patients. Organoid model experiments showed that IL-4/IL-13 selectively depleted CD73+CD104+ cells through STAT6 activation. This depletion triggers compensatory proliferation of CD73-basal cells, which exhibit defective differentiation and basal cell hyperplasia. Single-cell sequencing indicated that CD73+ cells have a low differentiation propensity and high regenerative capacity; their depletion disrupts epithelial homeostasis. These findings clarify the molecular pathways driving basal zone hyperplasia in EoE. 69
Furthermore, esophageal epithelial autophagy protects against the pathological changes of EoE Whelan et al. showed that inflammatory stimuli (TNF-α, IL-13) activate autophagy in esophageal epithelial cells via ROS-dependent mechanisms, based on mouse EoE and organoid models. Autophagy controls oxidative stress and preserves cellular homeostasis; its inhibition worsens oxidative injury and basal cell hyperplasia, which confirms autophagy as a key protective response to inflammation. 99
Collectively, esophageal organoid models have advanced the mechanistic understanding of EoE and other inflammatory esophageal disorders by preserving epithelial differentiation hierarchies and barrier-associated phenotypes in a 3D culture system. They recapitulate key disease-relevant histological and functional features and enable precise dissection of epithelial-intrinsic responses to inflammatory mediators, including effects on barrier integrity, lineage specification, and regenerative homeostasis—functional advantages that are limited in 2D epithelial models due to reduced differentiation maturity and structural complexity. Although epithelial organoids do not fully capture the immune complexity of EoE, they provide a well-controlled platform for dissecting epithelial-intrinsic responses to inflammatory cues. The translational relevance of these insights is further enhanced when organoid-derived findings are integrated with animal studies or immune/stromal co-culture approaches.
Tumor-related diseases
Esophageal squamous cell carcinoma
Esophageal squamous cell carcinoma (ESCC) is the main histologic subtype of esophageal cancer and originates from mucosal squamous epithelial cells. Its development involves both internal and external factors, including gene mutations, chromosomal copy number changes, tumor microenvironment alterations and DNA repair defects. Moreover, repeated mucosal injury and inflammatory stimulation are major external contributors to ESCC carcinogenesis.
ESCC organoid disease modeling is primarily achieved through two approaches. The first approach directly employs tumor organoids to investigate ESCC. Tumor organoids are generated either from patient-derived ESCC samples or from mouse tumors induced by carcinogens or engineered mutations. Isolated cancer cells are cultured in 3D to form organoids that recapitulate tumor architecture, molecular traits, and drug responses, serving as key platforms for mechanistic research and personalized therapy. 100 Importantly, patient-derived organoids maintain original mutational profiles and preserve cellular diversity, capturing tumor heterogeneity. Zheng et al. used genetically engineered mouse organoids by activating KrasG12D and deleting Trp53 via adenoviral Cre recombinase. This model captures early ESCC pathologic features and provides a robust resource for studying disease mechanisms and drug screening. 15
Another approach uses normal esophageal organoids as models for ESCC. Specifically, carcinogenesis is induced in normal organoids via gene editing or carcinogen exposure, which recapitulates ESCC initiation and progression. These models effectively capture tumor heterogeneity and dynamics, allowing detailed study of ESCC pathogenesis. Ko et al. used CRISPR/Cas9 to knock out TP53, CDKN2A, and NOTCH1 in mouse organoids, demonstrating that triple gene loss (PCN model) significantly drives tumorigenic transformation of esophageal epithelium. This model identified critical oncogenes involved in ESCC progression and also uncovered immune evasion characteristics. 73
ESCC typically develops from normal epithelium, progressing through low- and high-grade intraepithelial neoplasia before becoming invasive carcinoma. Zhang et al. developed organoids from normal and neoplastic human esophageal tissue. They demonstrated that ESCC organoids remodel the microenvironment and induce cancer-associated fibroblast (CAF) transformation, providing a platform to support therapeutic research targeting tumor–stroma interactions. 71 Similarly, Chen et al. used organoids derived from various ESCC stages to investigate epithelium–fibroblast crosstalk, highlighting ANXA1–FPR2 interaction as an essential homeostatic “brake.” 43 Liu et al. established an esophageal organoid model that recapitulates the full spectrum of tumorigenesis, from normal epithelium to invasive carcinoma. By integrating multiplex immunofluorescence with molecular profiling, they were the first to visualize spatial disruption in tissue architecture and dynamic shifts in key biomarkers (e.g. PD-L1 and Ki-67) within patient-matched organoids. This platform provides robust in vitro evidence linking phenotypic evolution to molecular signatures, elucidating the stepwise progression of precancerous lesions into malignancy. 56
Esophageal organoids are robust models for studying ESCC oncogenesis and progression. They allow detailed investigation of molecular mechanisms involved in tumor growth, cell differentiation, apoptosis, invasion, and metastasis. Wu et al. found that SOX2 in engineered mouse organoids induces endogenous retrovirus and double-stranded RNA expression, promoting tumor dependence on ADAR1 and revealing a novel cancer-driving pathway. 72 Tang et al. showed via ESCC organoids that Rab11-FIP1 hampers ZEB1 through the endocytic recycling pathway, preserving epithelial integrity and phenotype, impeding cell invasion, and restraining malignancy. 101 Du et al. used cell lines and patient-derived ESCC organoids to reveal that mTOR limits cancer stem cell properties by suppressing autophagy. 102 Yu et al. associated ΔNp63 with viral mimicry via retrotransposon activation in ESCC organoids and proposed a dual oncogenic process—epigenetic regulation and immune landscape remodeling. 103
Overall, ESCC pathogenesis is not driven by a solitary event but results from the synergy of multiple processes, including chromosomal instability, dysregulated cell fate, and epithelial-microenvironment interplay. By effectively preserving tumor genetic features and heterogeneity, ESCC organoid models offer a highly controllable system for elucidating oncogenic pathways, evolutionary trajectories, and resistance mechanisms. Integrating gene editing with functional perturbations, organoid studies have advanced our systemic understanding of ESCC molecular pathology, underpinning targeted therapy development and validating the translatability of therapeutic responses across multi-model frameworks.
Barrett’s esophagus
Barrett’s esophagus (BE) is a pathological condition characterized by the replacement of the normal distal esophageal stratified squamous epithelium with columnar epithelium. BE arises as a complication of gastroesophageal reflux disease, and it is the primary precursor lesion of esophageal adenocarcinoma (EAC). Its pathogenesis is multifaceted and includes gene regulatory changes, abnormal signaling pathway activity, and disrupted cellular differentiation.
In BE studies, organoid models derived from the gastroesophageal junction (GEJ), patients, and gene editing are crucial tools for disease modeling. BE involves lineage plasticity at the squamocolumnar junction and human-specific epithelial fate determinations—features that many animal models fail to fully recapitulate. 104 GEJ/BE organoids offer a superior, human-relevant platform for studying these processes. This system enables the dissection of epithelial-intrinsic transcriptional programs, epigenetic landscapes, and the effects of exogenous stimuli (e.g. acid and bile salts), providing crucial insights into disease mechanisms. Lian et al. constructed organoids from GEJ fundic progenitors and showed that p53 mutation drove neoplastic transformation toward atypia instead of metaplasia, underscoring p53′s pivotal role in BE cancer progression. 74 Similarly, Liu et al. generated APC-deficient BE organoids via CRISPR-Cas9, demonstrating the Wnt/β-catenin pathway activation as a key driver of tumorigenesis. This work first recapitulated malignant transformation of BE in 3D culture. Loss of APC led to nuclear β-catenin accumulation, upregulation of AXIN2 and c-MYC, increased proliferation, reduced apoptosis, and basement membrane disruption. These findings provide direct evidence of early BE oncogenesis and validate Wnt signaling activation as an independent tumor driver. 105 Zhang et al. treated BE organoids with acidic bile salts, which enhanced epithelial–mesenchymal plasticity in collagen I matrices and revealed mechanisms underlying subsquamous intestinal metaplasia formation in BE. 52
The cellular origins and mechanisms of BE are key research topics. Nowicki-Osuch et al. integrated single-cell transcriptomics, methylation lineage tracing, and functional validation in cardiac organoids. Their results show that BE molecular traits are dictated by c-MYC and HNF4A transcriptional programs. Crucially, they found EAC may arise from undifferentiated cells carrying BE molecular signatures, even in the absence of morphological features of BE. This insight identifies early transformation markers, thus challenging the classic idea that BE originates solely from squamous epithelium or submucosal glands. 75 Building on this, Jin et al. generated a BE organoid biobank from patients, reproducing BE molecular heterogeneity and demonstrating that the SOX2/CDX2 expression balance is pivotal for BE differentiation. Their findings reveal SOX2 as a central foregut epithelial regulator, and SOX2 is significantly diminished in BE development. Lineage tracing confirmed that certain BE glands derive directly from SOX2-deficient squamous cells, suggesting that fate conversion is one cellular origin of BE, though BE likely has multiple cellular sources. 76
Multiple signaling pathways govern malignant transformation in BE. Kunze et al. used BE organoids and L2-IL1B mice to show that Notch signaling drives EAC. It does so by activating NF-κB, inhibiting goblet cell differentiation, and expanding progenitor populations. 77 Correi et al. used humanized BE organoids to confirm that BMP2/4 inhibition eradicated BE metaplastic cells and stimulated squamous progenitor regeneration. BMP7 was found to promote squamous differentiation, demonstrating the dual role of BMP signaling in BE cell fate regulation. 106 De et al. showed that FOXF1 overexpression in NES-G4 organoids converts squamous spheres to glandular structures, substantiating the mechanisms of BE formation. 78 Sun et al. modeled organoids with p16 loss and KRASG12D activation, showing that p16 deficiency induces LGR5+ metaplasia while KRASG12D accelerates high-grade dysplasia. Together, their findings clarify the synergy between cell cycle dysregulation (G1/S transition) and MAPK activation in BE’s malignant progression, presenting new targets for early intervention. 107
Sahm et al. used L2-IL1B.mTERC-/- G2 mouse models together with 3D organoid culture to demonstrate that telomere shortening induces DNA damage and clonal selection, which accelerates BE malignancy. Telomere dysfunction boosts BE progenitor proliferation and survival, highlighting telomere crisis as crucial in tumor initiation and offering an experimental platform for early EAC research. Human BE cell telomere profiling showed goblet cells retain long telomeres, whereas columnar cells display marked shortening. This creates a heterogeneous “cellular soil” that favors malignant transformation, which is also recapitulated in organoids. 108
These studies support the idea that BE is not caused by a single pathway but results from cellular plasticity and reprograming of multiple epithelial lineages under specific genetic and environmental conditions. Organoid models offer a critical platform to study epithelial transcriptional and epigenetic programs, elucidating the molecular basis of lineage fate transitions. By reproducing metaplasia and early dysplasia, organoid research has advanced our understanding of BE pathogenesis and provided a framework for exploring genetic and environmental interactions in disease progression.
Esophageal adenocarcinoma
Esophageal adenocarcinoma (EAC) originates from distal esophageal glandular tissue. The incidence of EAC has sharply increased in recent years. Prognosis remains poor, which has fueled intense research interest in digestive oncology. EAC develops through multiple stages: chronic inflammation transforms the squamous epithelium into Barrett’s esophagus (BE), which then progresses from low- to high-grade dysplasia and finally to invasive adenocarcinoma. This progression is governed by genetic, epigenetic, and environmental contributors. Hallmarks of EAC include genomic instability, such as chromothripsis, amplicon rearrangement, and epigenetic dysregulation, including aberrant DNA methylation.109,110
Patient-derived EAC organoids now provide breakthrough platforms for modeling EAC disease. Derouet et al. generated EAC organoids from endoscopic biopsies that recapitulate key tumor molecular profiles, including approximately 60% SNV concordance and partial copy number alterations, and accurately mimic tissue architecture and drug responses. 59 Fan et al. standardized single-cell digestion and cryopreservation, which boosted EAC organoid reproducibility and facilitated the development of robust, efficient research platforms for EAC. 111
Researchers have clarified the molecular pathways involved in premalignant lesion evolution and carcinogenic mechanisms in EAC using BE tissue-derived organoids. Their findings reveal the progression from non-progressive BE to high-grade dysplasia and eventually invasive adenocarcinoma. Flis et al. showed that TLR2 signaling was essential during the transition from BE to early esophageal adenocarcinoma. Continuous activation of this pathway induces HMGB1 release and macrophage polarization, which promotes a tumor-supportive inflammatory microenvironment and enhances conditions that drive disease progression. 112 Barber et al. further characterized dynamic changes in caspase-1 expression across stages of BE. Caspase-1 facilitates a localized esophageal inflammatory microenvironment by regulating secretion of mediators like IL-1β and CXCL1. The caspase-1-dependent inflammatory response is closely linked with BE maintenance and early tumor development, which provides new mechanistic evidence for inflammation-driven esophageal carcinogenesis. 113
The development of EAC is characterized by widespread genomic aberrations and disruption of signaling cascades. Frankell et al. conducted whole-genome and transcriptome sequencing of 551 EAC cases. They systematically constructed a comprehensive landscape of driver events in esophageal adenocarcinoma (EAC). They identified 76 driver genes, including TP53 and SMAD4. More than 50% of cases harbored genomic alterations, such as CCND1 and CCND3 amplifications or KRAS activation, which conferred sensitivity to CDK4/6 inhibitors. They used patient-derived EAC organoids to further confirm the efficacy of these inhibitors and recapitulated the resistance phenotype mediated by CCNE1 amplification, thereby providing critical preclinical functional evidence that supports genome-guided precision therapy for EAC. 79 Scott et al. integrated genomic profiling with patient-derived organoid models and systematically revealed that EAC polyploidy originated from mitotic slippage caused by Ndc80/Dsn1 imbalance. Organoids not only validated the two-dimensional findings but also directly linked copy number variations to functional abnormalities, thereby establishing a coherent molecular-to-clinical evidence chain. 114
Ijaz et al. combined haplotype-resolved chromosomal assembly with EAC organoid models. This approach revealed complex structural rearrangements induced by chromothripsis, such as single chromosomes, containing more than 900 structural variants. Multi-omics analyses further showed that these rearrangements promoted tumor progression by reorganizing three-dimensional genome architecture, including TAD boundary remodeling, and by altering the local epigenetic landscape, as exemplified by the enrichment of differential peaks near structural variants. 115
Jammula et al. integrated DNA methylation, genomic, and transcriptomic data. Using this integration, they classified Barrett’s esophagus (BE) and EAC into four molecular subtypes. The organoid models reproduced both the molecular features and heterogeneity of these subtypes, while functional assays identified key epigenetic regulatory mechanisms. Alterations in the Wnt/β-catenin signaling pathway were detected in specific subtypes. Moreover, distinct pathway activation patterns were identified: subtype 1 exhibited enrichment of cell cycle processes, subtype 2 showed metabolic reprograming, subtype 3 demonstrated immune pathway activation, and subtype 4 was associated with genomic instability. Collectively, these findings provide new insights into the molecular mechanisms underlying EAC heterogeneity. 80
Ng et al. systematically integrated genomic profiles from 710 EAC cases with organoid models. This approach provided mechanistic insight into the amplification of the oncogenes ERBB2 and MYC. Two major amplification pathways were characterized: extrachromosomal DNA (ecDNA) formation and breakage-fusion-bridge (BFB) cycles. Amplified genomic regions were enriched with H3K27ac-marked enhancer elements, and these variants originated during precancerous stages. Furthermore, single-cell sequencing established that ecDNA drives clonal dynamics in tumors. This discovery highlights the functional significance of structural genome variation in the initiation and progression of EAC. 81
van der Zalm et al. identified the pluripotency factor NANOG as a pivotal regulator of mesenchymal plasticity in EAC through integrative multi-omics analyses and functional validation. They also found that NANOG expression levels strongly correlated with patient prognosis. The study used cell lines, organoids, and mouse models to systematically define the mechanisms underlying resistance-associated plasticity in EAC and establish a platform for designing personalized therapies. Moreover, drug sensitivity assays in organoids showed that niclosamide significantly enhanced the radiosensitivity, providing a complementary ex vivo platform linking NANOG-driven plasticity mechanisms to clinically relevant combined chemoradiotherapy strategies. 82
In summary, the pathogenesis and progression of EAC constitute a dynamic evolutionary process driven by genomic instability, epigenetic remodeling, and clonal selection. PDOs largely preserve the molecular features and evolutionary trajectories of primary tumors, providing a powerful experimental platform to dissect the mechanisms underlying structural variation formation, tumor heterogeneity maintenance, and cancer stemness regulation. Through the integration of multi-omics analyses and functional validation, organoid-based models offer unique advantages in identifying key oncogenic events and actionable therapeutic targets in EAC, thereby establishing a robust foundation for the translational evaluation of treatment strategies within integrated multi-model experimental systems.
Drug screening and personalized medicine
The treatment of esophageal cancer has long faced several challenges. These include chemotherapy resistance, limited options for targeted therapy, and uncertain efficacy of immunotherapy. Esophageal organoids partially retain tumor heterogeneity. They have been widely used for drug screening and personalized therapy research. By studying esophageal tumor organoids, we can systematically evaluate drug sensitivity and resistance mechanisms, identify novel therapeutic targets (Table 3), and provide critical evidence to support clinical decision-making and precision medicine. However, current standard organoid culture systems have inherent limitations, such as the absence of functional immune cells (e.g. T cells and tumor-associated macrophages), stromal components (e.g. tumor-associated fibroblasts), and vascular networks. In addition, selective culture conditions may lead to the loss of specific subclonal populations. Therefore, these limitations should be carefully considered when interpreting the drug screening results.
Research progress on esophageal cancer organoids for drug sensitivity testing.

Note.
“a” (Level 1) - Mechanistic Discovery: Focuses on identification of intrinsic vulnerabilities or novel biomarkers within organoids.
“b” (Level 2) - Clinical Safety Guidance: Involves PDO results that accurately recapitulate or explain known clinical trial phenomena.
“c” (Level 3) - Clinical Outcome Validation: Demonstrates the potential consistency between in vitro sensitivity and clinical outcomes.
Esophageal squamous cell carcinoma
Chemoresistance mechanisms in ESCC significantly limit chemotherapy efficacy. Fuchino et al. were the first to establish paired organoid models of ESCC, representing samples taken before and after neoadjuvant chemotherapy (PreNAC-O/PostNAC-O). Through functional experiments, they demonstrated how residual cancer cells adapt and change over time. This study utilized the ability of organoids to preserve tumor heterogeneity and microenvironmental responses. The results showed that residual cancer cells after chemotherapy acquire increased tumorigenic capacity through epithelial–mesenchymal transition (EMT) and hypoxia pathways, rather than arising from pre-existing resistant clones. 83 This “paired-sample” strategy offers a research framework for studying therapy-induced dynamic evolution. It warrants broad application in future drug screening.
Targeted therapeutics have greatly improved treatment selectivity and efficacy by revealing mechanisms in specific molecular subtypes. Lin et al. utilized patient-derived organoid (PDO) models to validate the enhanced sensitivity of tumors with low TACC2 expression to the CDK inhibitor dinaciclib, which showed a 78% decrease in IC50. In addition, parallel studies using murine model studies confirmed that combination therapy with CDK inhibitors and TACC2 deficiency significantly enhances therapeutic efficacy. This led to an 89% decrease in tumor size. These results clarify the molecular mechanisms of the TACC2-CDKN1A regulatory axis and offer strong theoretical and experimental support for precision therapy of ESCC based on synthetic lethality. 116
Nakagawa et al. established an ESCC organoid biobank from 24 patients, revealing that chromatin accessibility at NRF2 pathway loci serves as a key epigenetic mechanism driving chemoresistance. Large-scale screening identified fedratinib, a JAK2 inhibitor, as a potent agent against resistant organoids. Interestingly, fedratinib functions through NRF2-independent pathways, potentially by inhibiting BRD4. This study not only substantiates the utility of organoid models in drug discovery but also proposes biomarkers (e.g. ALDH3A1) to guide precision treatment for refractory ESCC. 44
Due to frequent monotherapy resistance in ESCC, combination strategies targeting convergent resistance nodes are pivotal for overcoming failure and enhancing efficacy. Spender et al. demonstrated in cell lines, CDX, and PDOs that diverse resistance mechanisms—including PDGFRβ dependence, IGF1R activation, and EMT—all converge on sustained AKT/mTOR signaling in EGFR-driven ESCC. AKT/mTOR and EGFR co-inhibition exhibited pronounced synergy in PDO models. Despite dose-dependent toxicities, dosing optimization can balance efficacy and safety, offering a clinical strategy for EGFR inhibitor resistance. 120 However, in vitro organoid models cannot fully replicate the in vivo absorption, distribution, metabolism, and excretion of drugs. Therefore, the toxicity profiles and optimal dose ratios of combination therapies still require validation in prospective clinical trials.
Collectively, the findings of Spender et al. and Nakagawa et al. demonstrate that distinct resistance mechanisms (AKT/mTOR signaling vs NRF2-driven epigenetic alterations) may require different combination strategies. These results underscore the value of organoid models in identifying patient-specific resistance mechanisms.
Radiation therapy is a crucial approach in treating ESCC. Its efficacy largely depends on the sensitivity of tumor cells to radiation. Its efficacy in ESCC largely depends on the sensitivity of tumor cells to radiation. Carswell et al. used mouse-derived 3D esophageal organoids to investigate the damaging effects of varying radiation quality and dose on esophageal epithelium. They revealed key radiation-induced cytological events, such as thickening of the basal-like cell layer, reduced involucrin expression indicating differentiation abnormalities, and activation of the p38 MAPK pathway. These findings elucidate the molecular basis of radiotherapy-associated esophageal tissue lesions. They also provide crucial experimental evidence and potential targets to better understand radiation sensitivity mechanisms and to develop combined radiotherapy–drug treatment intervention. 121 Notably, the study used organoids derived from normal esophageal epithelium, whose radiation responses may differ from those of tumor-derived organoids.
Zhang and Wang established a comprehensive platform linking patient tumor samples with patient-derived xenograft (PDX) and PDX-derived organoid (PDXO) models. This standardized platform enables the evaluation of radiotherapy sensitivity in ESCC. By integrating in vivo and in vitro approaches, the platform effectively recapitulates tumor heterogeneity and radiotherapy response. However, its prolonged establishment time and the technical complexity of surgical procedures limit its applicability in point-of-care clinical decision-making. 122
Studies investigating immune co-cultures of esophageal cancer organoids remain in their infancy, and no standardized platform is currently available to fully recapitulate the tumor immune microenvironment. Jiang et al. established murine and human oral and esophageal organoid models to study tumor immune responses. They developed a novel co-culture system combining tumor organoids with autologous CD8+ T lymphocytes. This system enables quantitative assessment of T cell–mediated cytotoxicity by flow cytometric detection of cell death markers and provides an in vitro platform for evaluating CD8+ T cell–driven antitumor activity. Importantly, this co-culture system represents a significant advance over purely epithelial organoid models in immunotherapy research. 12 However, as a reductionist approach, it does not fully recapitulate the complex immunosuppressive tumor microenvironment characteristic of esophageal cancer. Consequently, the ability of this model to predict clinical responses to immune checkpoint inhibitors requires rigorous prospective clinical validation.
In summary, ESCC organoids provide a physiologically relevant platform for drug screening and for studying key biological processes, including chemoresistance, targeted therapy, combination strategies, radiosensitivity, and tumor-immune interactions. They have enabled the evaluation of precision therapeutic strategies based on synthetic lethality and pathway interactions and have generated important experimental evidence supporting radiosensitization and immunotherapy. At the same time, current organoid systems remain limited by the absence of a fully reconstituted tumor microenvironment—particularly immune components—as well as by incomplete modeling of in vivo pharmacokinetics and relatively long establishment times. These limitations restrict their applicability in real-time clinical decision-making. Consequently, while organoid-based assays are highly predictive for tumor cell–intrinsic resistance and direct cytotoxic agents, results regarding therapies dependent on immune or vascular-oxygenation contexts warrant cautious interpretation.
Esophageal adenocarcinoma
Esophageal adenocarcinoma (EAC) is a highly heterogeneous digestive tract malignancy. Clinical treatment outcomes vary significantly due to molecular and biological differences among patients. Therefore, there is an urgent need for more precise drug screening platforms and personalized treatment strategies.
Despite the limited availability of esophageal adenocarcinoma (EAC) organoid models, Li et al. successfully established 10 organoid lines from 32 patients, representing a success rate of 31%. Nine of these lines sustained long-term culture while retaining the molecular profiles and dominant clonal architecture of the primary tumors. Multi-timepoint sequencing further revealed that serial passaging exerted selective pressures in vitro, leading to subclonal loss and dynamic clonal evolution. In particular, subclones harboring driver mutations, such as RNF213, showed proliferative advantages during culture, thereby highlighting the utility of organoid models for studying clonal dynamics and the evolution of therapeutic resistance. 13
Chemoresistance to conventional platinum-based agents has been shown to involve intricate mechanisms. Ballout et al. demonstrated that suppression of SMAD3 phosphorylation or expression significantly enhances oxaliplatin sensitivity in EAC models to oxaliplatin. Mechanistically, this effect is mediated by relief of SMAD3-induced PP2A inhibition, which subsequently blocks ATM-dependent DNA repair. This study suggests that targeting the SMAD3-PP2A-ATM axis may be a novel strategy to enhance the efficacy of conventional chemotherapy. 117 In addition, Stevenson et al. showed that the AKT inhibitor ALM301 increases chemotherapy sensitivity by blocking the PI3K/AKT pathway. Meanwhile, the IAP antagonist BV6 overcomes resistance to AKT inhibition. This synergy was validated in patient-derived organoids, including the resistant CAM401 line, supporting a combined targeted therapeutic approach. 84
Furthermore, Dings et al. found that radiotherapy-induced ROS activates ESRRA in EAC, which then upregulates mitochondrial biogenesis and oxidative phosphorylation. This study elucidates a novel mechanism underlying EAC radiotherapy resistance. It should be noted that this model does not capture human pharmacokinetics, drug distribution, or systemic toxicity. Crucially, the risks of ESRRA inhibition in metabolic hubs, particularly cardiac and hepatic tissues, remain poorly characterized and fall outside the scope of this model. 123
Organoid models have also shown great value in targeted drug screening and efficacy prediction. Lin et al. used 3D tumor spheroid models of EAC cell lines, and showed that cardiac glycosides suppress EAC cell proliferation by inhibiting the Na+/K+ ATPase–MAP2K6 (MKK6)–SOX9 signaling pathway both in vitro and in vivo. This work provides mechanistic support for targeting this pathway in EAC treatment. 124
Concurrently, Smyth et al. integrated organoid drug sensitivity results with clinical trial data, revealing that patients with EGFR amplification experienced worse outcomes when treated with panitumumab combined with anthracycline chemotherapy. Organoid experiments showed that this adverse effect results from an antagonistic interaction between anti-EGFR drugs and anthracycline chemotherapy. This study provides a rare example of confirming the efficacy of organoid-based combination therapy with actual clinical treatment outcomes. Such “clinical-to-organoid” validation approaches remain relatively uncommon in organoid research and oncology. 118
Metabolic reprograming is a key mechanism of tumor drug resistance. Recent studies using organoid models have further clarified this process. Su et al. showed that lactate suppresses the growth of patient-derived EAC organoids through modulation of intratumoral NADH/NAD+ redox status. These findings indicate that shifts in metabolic pathways can affect drug response and offer rationale for metabolism-targeted therapeutic development. 125 McNamee et al. found that AMPK activation promotes both survival and resistance to chemotherapy and radiotherapy in EAC, mainly by enhancing oxidative metabolism. Combined treatment with AMPK inhibitors markedly improved therapeutic efficacy in organoids, supporting metabolic intervention strategies. 85 Ju et al. demonstrated across models that AKR1C3 inhibits TRIM21-mediated ubiquitination of HSPA5. This inhibition stabilizes the HSPA5/GPX4 axis and limits ferroptosis, thereby promoting radioresistance in EAC. These results reveal cross-talk between AKR1C3-dependent metabolic reprograming and ferroptosis and confirm that targeting this axis can enhance radiotherapy efficacy. 126 These findings indicate that metabolic reprograming can affect drug response and offer rationale for metabolism-targeted therapeutic development.
Bolger et al. utilized organoid models derived from EAC patients. They identified subgroups sensitive to cisplatin chemotherapy and others unresponsive to conventional chemotherapy but sensitive to alternatives such as irinotecan. This finding provides important preclinical evidence for personalized treatment of esophageal cancer, as the sensitivity to cisplatin (IC50) in patient-derived organoid models significantly correlates with the pathological regression degree in patients undergoing neoadjuvant therapy and the radiological response in metastatic patients. 119 However, the use of organoid models remains in the research phase and has not yet been applied to clinical decision-making. 119 Despite these promising results, key clinical application parameters, such as organoid establishment success rates and required testing time, were not reported. And the study sample size was limited. Larger-scale prospective studies are needed to validate its clinical value.
Overall, studies of EAC organoids initiated correlative validation with clinical data at an early stage, as exemplified by Li et al. and Bolger et al., reflecting a strong translational orientation. In this context, the retrospective organoid analysis of the REAL3 trial by Smyth et al. represents a rare example in which organoid-based findings were directly linked to actual clinical outcomes. In parallel, metabolic reprograming has been more extensively investigated in EAC organoid models, likely reflecting the unique Barrett’s esophagus–dysplasia–carcinogenesis continuum characteristic of EAC. Together, these studies highlight the utility of patient-derived organoids for interrogating tumor heterogeneity, clonal evolution, and therapy resistance mechanisms. Nevertheless, similar to ESCC, EAC organoid models face important limitations, including incomplete preservation of tumor heterogeneity and the lack of an immune microenvironment. These constraints warrant careful consideration when extrapolating organoid-based conclusions to in vivo settings.
Regenerative medicine
Esophageal diseases include various pathological conditions. These involve structural anomalies, motility dysfunction, inflammation, and tumors. Some patients remain asymptomatic or experience mild symptoms with little health implications, while others develop dysphagia or life-threatening complications requiring esophagectomy. However, esophagectomy results in considerable lifestyle disruption and increases the risk of secondary diseases. Compared to other organs, the relative simplicity of esophageal anatomy reduces barriers to regenerative medicine applications in esophageal repair.
To restore feeding capacity after partial esophagectomy, clinical protocols commonly use autologous tissue such as stomach, colon, or jejunum for esophageal reconstruction. Although these techniques repair esophageal defects, they cause significant trauma and complications that worsen clinical outcomes and patient quality of life.
In preclinical studies, human intestinal organoids (HIOs) were transplanted into the mesentery of immunodeficient mice. This procedure resulted in structural integration and vascularization. These constructs displayed tissue organization and stem cell activity that are comparable to human intestine and supported surgical procedures such as anastomosis. However, immune interactions have not been evaluated because of the immunodeficient host, and functional maturation, such as absorption and peristalsis, requires further validation. These findings establish a technical foundation for intestinal regenerative medicine; however, therapeutic efficacy and safety must be validated in larger animal or disease models, such as larger species or increased sample sizes. Although intestinal organoid technology inspires new approaches for managing esophageal disease, esophageal organoids themselves have not yet been applied in regenerative therapy. 86
Spurrier et al. pioneered the esophageal organoid unit (EOU) model. They used enzymatic digestion of neonatal mouse or human tissue to isolate multicellular clusters comprising epithelial and mesenchymal progenitors. Neuromuscular cells were not required in co-culture for network formation. Under Matrigel culture, the EOUs spontaneously formed polarized 3D structures with an epithelial layer showing a gradient of basal (PCNA+CK14+) and suprabasal differentiated (CK4+CK13+) cells. Notably, during culture, EOUs developed a functional neuromuscular network. Immunofluorescence revealed that Tuj-1+ neural fibers and SMA+ smooth muscle cells formed interconnected networks, enabling spontaneous contractions resembling esophageal peristalsis. 87
Subsequent experiments confirmed that EOUs expanded in vitro and then implanted on PGA/PLLA collagen scaffolds generated tissue-engineered esophagus (TEE) in vivo. This engineered tissue contained complete epithelial, muscle, and neural components. The PGA/PLLA collagen scaffold comprises a three-component composite tubular structure, featuring polyglycolic acid (PGA) nonwoven felt as the porous scaffold, poly-l-lactic acid (PLLA) solution as the shaping binder, and type I collagen as the bioactive coating. This construct provides temporary mechanical support and creates an optimal microenvironment that promotes EOU survival, proliferation, and self-assembly into a fully structured neo-organ with epithelial, muscular, and neural components. Both PGA and PLLA are biodegradable and bioresorbable polymers approved by the US FDA for clinical applications. These findings illustrate the utility of EOUs as a cell source for esophageal regeneration and highlight the strong self-organization ability of esophageal progenitors, which can produce functional engineered tissue even when the initial cell arrangement is disrupted. This strategy provides a new clinical option for esophageal atresia and cancer. Expansion and scaffold implantation of autologous EOUs may facilitate functional esophageal reconstruction. 87
In summary, esophageal organoids serve as a key bridge connecting fundamental research with clinical application (Figure 3). They improve the modeling of esophageal diseases, deepen our understanding of their mechanisms, advance novel drug development, and facilitate the formulation of personalized treatment strategies. The prospects for their application are expected to expand considerably in the fields of precision medicine and regenerative medicine.

Schematic diagram illustrating the applications of human esophageal organoids: (A) mechanism of esophageal development—by simulating signaling pathways in vitro, from the gastrula stage to esophageal development; (B) disease modeling and mechanistic research—employing gene editing technologies to generate disease models; (C) drug screening and personalized medicine—utilizing high-throughput drug screening to guide precision therapy; and (D) regenerative medicine—aiming toward the future in vivo transplantation of organoids.
Challenges and countermeasures
Despite substantial advances in disease modeling and drug screening, the clinical translation of esophageal organoid technology remains impeded by persistent technical bottlenecks. Critical gaps in standardization protocols, microenvironment recapitulation, and genomic stability collectively compromise their transition from experimental tools to clinical decision-support systems. This section systematically dissects the roots of core challenges and proposes targeted solutions through interdisciplinary integration, thereby paving the way for precision esophageal medicine.
Standardization deficiency
Current esophageal organoid cultivation lacks unified protocols and quality criteria. Significant inter-laboratory variations in medium composition, matrix selection, and culture conditions lead to batch-dependent heterogeneity in organoid morphology, functionality, and drug sensitivity, severely compromising experimental reliability and cross-platform comparability. 127 Furthermore, reliance on costly reagents (e.g. growth factors/Matrigel) and complex procedures hinders technical standardization and adoption.
To address these challenges, standardized culture systems must be established alongside cost optimization. Research institutions should collaboratively develop comprehensive standard operating procedures (SOPs)—modeled after Lee et al.′s intestinal organoid standards 128 —covering cell sourcing and cultivation, as well as quality metrics. In detail, human adult stem cells (hASCs) are cultured using a three-dimensional (3D) protocol that supplements R-spondin 1 (Rspo1) and epidermal growth factor (EGF) for proliferative and functional maintenance. In contrast, human pluripotent stem cells (hPSCs) undergo directed differentiation from definitive endoderm to hindgut, thereby mimicking in vivo development. Comprehensive quality assessment includes morphological identification ensuring that essential cell types constitute at least 30%, post-cryopreservation cell viability of at least 50%, as well as functional activity and genetic stability verification. 128 This approach aims to establish highly reproducible, clinically applicable culture systems.
Notably, organoid standardization is progressing at both national and international levels. In China, a series of organoid-related standards, including T/CMBA 017-2022 and T/CSCB 0005-2022, have been released, covering gastrointestinal, lung, and brain organoids. 129 In parallel, the International Organization for Standardization (ISO/TC 276) has initiated the development of international organoid standards. 129 However, dedicated criteria for esophageal organoids remain insufficient, representing a major bottleneck for further methodological and clinical advancement.
Advancing the standardization of esophageal organoids requires the definition of minimal quality and performance benchmarks. These include reproducible thresholds for culture success rates (approximately 60–70% for ESCC organoids), 130 intra- and inter-patient consistency of drug response profiles, and validation metrics aligned with clinical endpoints such as treatment response and survival. In addition, quality criteria should ensure faithful reconstruction of the multilayered squamous epithelial architecture, including basal-to-superficial differentiation gradients and layer-specific marker expression (e.g. p63/CK14 in basal layers and CK13 in superficial layers). 9 Functional validation, such as barrier integrity, responses to pathological stimuli, and therapeutic sensitivity, is also required to establish clinically relevant organoid models.
Multicenter clinical validation represents a critical step toward standardization and clinical translation. For example, Shen et al. developed a standardized platform for ESCC organoid culture and drug sensitivity testing, achieving an overall culture success rate of 61.8% (34/55). In this cohort, organoid-based drug sensitivity predictions were concordant with clinical chemotherapy responses in 83.33% (10/12) of evaluable cases. 130 Similarly, Shao et al. proposed a two-step drug screening strategy that enables rapid identification of chemotherapy resistance and selection of alternative effective agents. Collectively, these studies provide practical reference frameworks for the standardized and clinically oriented application of esophageal organoids. 131
Cost control and technological accessibility are also important concerns. Actively developing low-cost substitutes and new synthetic extracellular matrices is a key strategy. For instance, Takahashi et al. demonstrated that L-WRNH cell-conditioned medium can replace costly recombinant proteins, reducing costs by approximately 100-fold, and simultaneously has no effect on the proliferation or function of the organoid. 132 L-WRNH cells are engineered mouse L cells that exogenously express HGF, Wnt3a, R-spondin1, and Noggin, which together provide the key microenvironmental factors required for organoid culture. Collagen gel is a viable substitute for Matrigel and can be combined with WRNH-conditioned media for organoid culture. Furthermore, researchers are exploring synthetic matrices with defined compositions and tunable properties—such as polyethylene glycol-based hydrogels for bile duct cysts. 133 These studies provide direction for establishing standardized, low-cost organoid systems and lay the groundwork for large-scale organoid production.
In summary, it is essential to systematically address the current obstacles to the standardization and generalization of esophageal organoids. We can achieve this by establishing uniform quality standards, defining esophagus-specific evaluation systems, promoting multi-center clinical validation studies, and developing low-cost culture protocols for disease modeling, drug screening, and clinical applications.
Genomic instability
During long-term culture, organoids experience genomic instability characterized mainly by chromosomal copy number variations (CNVs). Additionally, clonal evolution occurs, which includes accumulated gene mutations and increased subclonal heterogeneity. This phenomenon is especially pronounced in tumor-derived organoids, compromising their genetic fidelity and undermining the reliability of drug response predictions. For example, in head and neck cancer organoids, serial passaging can lead to significant clonal evolution and genetic drift, altering their sensitivity to chemotherapy and radiotherapy. 134
Esophageal adenocarcinoma (EAC) organoids undergo clonal evolution during early passages. However, they preserve overall genomic integrity. Their copy number variations and structural rearrangements closely resemble those of primary tumors. Key driver gene mutations (e.g. TP53, CDKN2A, PIK3CA) remain stable, and whole-genome sequencing shows that the mutation spectrum has no substantial changes during long-term culture. Clonal selection primarily occurs during the initial passage. Subsequent genomic variation increases by less than 25% compared to initial levels. Importantly, even after 6 months of culture, these changes do not affect the core driver genes. 13
To decrease risks of genomic instability, the following measurements are used. These measurements include controlling genetic drift by limiting passage numbers, regularly monitoring mutation dynamics via low-pass whole-genome sequencing, establishing cryopreserved cell banks from early passages, and confirming cell identity by performing short tandem repeat (STR) analysis to prevent cross-contamination.
For esophageal organoids, we recommend implementing a multi-level genomic monitoring system. For example, ESCC organoids, characterized by high mutation burden and a complex genome, should undergo targeted sequencing every 5–10 passages. 44 Monitoring should focus on the stability of driver genes such as TP53, NOTCH1, NFE2L2, and PIK3CA, 135 and the amplification status of chromosomal regions such as 3q and 8q. Barrett’s esophagus organoids should be monitored for loss of heterozygosity (LOH) at 17p and 9p and whole-genome duplication events. 136 For EAC organoids, besides tracking typical driver gene mutations, special attention will be given to amplifications in RTK pathway genes (e.g. ERBB2) and the presence of extrachromosomal DNA (ecDNA). 81 For all esophageal organoid types, it is recommended to establish a master cell bank at the third passage and to perform STR identity verification prior to use.
From a clinical translation perspective, the key is not only to minimize clonal drift but also to confirm the extent to which genomic evolution does not affect the accuracy of organoid-based drug sensitivity predictions. For instance, Shao et al. demonstrated that drug responses in ESCC organoids showed good concordance with actual patient outcomes within an approximately 30-day clinical decision window, suggesting that genomic changes during short-term culture do not yet interfere with predictive efficacy. However, for applications requiring long-term expansion, such as constructing drug screening libraries or conducting mechanistic studies, stricter genomic monitoring strategies are necessary. 131 Such strategies include careful recording of passage numbers to ensure the reliability and reproducibility of results.
In summary, establishing monitoring protocols specific to organoid types, standardizing cell bank management, and differentiating quality control requirements for short-term clinical testing versus long-term research-all of these can effectively decrease genomic instability in esophageal organoids. This approach enhances the credibility and translational potential of esophageal organoids as disease models and predictive tools.
Inadequate microenvironment recapitulation
A fundamental goal of organoid technology is to construct structures that closely mimic native tissues. However, current esophageal organoid models remain limited in their ability to faithfully reproduce the native tumor microenvironment, particularly with respect to immune complexity and intratumoral heterogeneity. The absence of a functional immune microenvironment represents a major bottleneck in existing organoid systems. Most culture platforms lack key immune components, including T lymphocytes, natural killer (NK) cells, tumor-associated macrophages (TAMs), and myeloid-derived suppressor cells (MDSCs), all of which play critical roles in tumor progression, immune evasion, and therapeutic response. As a result, organoid models without immune components are poorly suited for immunotherapy research, as tumor–immune interactions are essential for determining clinically relevant treatment outcomes. In addition, current organoid models show substantial deficiencies in recapitulating the tumor stroma. Conventional systems typically lack cancer-associated fibroblasts (CAFs), endothelial cells, and a physiologically relevant extracellular matrix (ECM). These stromal components regulate tumor behavior through paracrine signaling and form physical barriers that restrict drug penetration, thereby contributing to therapeutic resistance. Moreover, the absence of functional vascular networks prevents the establishment of realistic drug distribution dynamics and intratumoral hypoxic gradients, further limiting the predictive value of organoid models for chemotherapeutic and radiotherapeutic responses.
The establishment and long-term culture of organoids can introduce selection biases, leading to the preferential expansion or loss of tumor subclones with different proliferative capacities. This process may compromise the faithful representation of intratumoral heterogeneity and reduce the accuracy of drug sensitivity predictions. In addition, substantial variability in organoid establishment success rates across patient cohorts and tumor subtypes can introduce systematic sampling bias, thereby limiting the ability of organoid biobanks to capture the full spectrum of disease diversity.
Microfluidic organoid-on-a-chip technology provides a transformative method to address deficiencies in mimicking the microenvironment. Sharpe et al. developed an assembled EAC model by co-culturing organoids with CAFs at a 2:1 ratio. This model successfully maintained patient-specific CAF phenotypes and revealed preliminarily their functional and spatial heterogeneity, providing a valuable 3D platform for studying stromal-mediated tumor biology. 137 Pal et al. utilized an Emulate microfluidic platform to develop a patient-specific EAC organ-chip model. In this system, patient-derived tumor cells grow within the top “epithelial” channel, while matched CAFs reside within the bottom “stromal” channel separated by a porous membrane. This design simulates the in vivo tumor-stroma interface. These efforts mark significant progress toward sophisticated co-cultures. 138 Future developments should aim to produce more valuable chips integrating immune cells and vascular networks for enhancing the predictive value of these chips in mechanism studies and therapeutic screening.
Dynamic functional gaps
The challenge of accurately simulating functional properties remains a core bottleneck that hinders applications of esophageal organoids in translational medicine. Current models have notable limitations, which mainly concern two aspects: mechanical dynamics and chemical microenvironments.
The esophagus is a muscular tube controlled by neuromuscular coordination. Its mechanical functions, such as sphincter relaxation and coordinated peristalsis, rely on intact innervation and functional smooth muscle units. 139 However, most existing organoids are mainly composed of epithelium and lack functional neuronal and muscular components. The absence of these components makes it difficult to mimic mechanical function and to study esophageal motility disorders like achalasia in depth. 52
To address this limitation, developing neuro-smooth muscle units with functional output has emerged as a key research focus. Although esophageal organoids have not yet achieved a mature in vitro peristalsis model, relevant differentiation research provides a reference path. Studies on enteric nervous system differentiation from human pluripotent stem cells (hPSCs) show that modulating specific signaling pathways (e.g. PDGFR) directly induces efficient differentiation of nitric oxideergic neurons (NOS1⁺), which mediate smooth muscle relaxation and improve motor function of the stomach and intestine in transplantation experiments. 140 This suggests that three-dimensional co-culture of these functional neurons with esophageal smooth muscle cells holds promise for building pathology-specific functional units. This approach offers a new paradigm for the in vitro modeling of achalasia.
As independently cultured units, esophageal organoids lack functional connections to adjacent organs in vivo (e.g. stomach, pharynx). This isolation hinders precise recapitulation of pathological exposures in GERD. These exposures include periodic reflux of gastric acid and bile, fluid shear stress, and synergistic damage caused by multi-component refluxates (e.g. acid, bile, enzymes). Furthermore, the lack of a standardized exposure-response assessment system undermines the physiological relevance and data comparability of the models. A promising strategy to overcome this bottleneck is to combine the biological complexity of organoids with engineered platforms. Specific technical approaches include establishing two-dimensional polarized epithelial monolayers derived from organoids using transwell or air-liquid interface culture systems. These enable stable and controllable apical chemical exposure and functional assessment; reconstructing near-physiological dynamic exposure environments by incorporating microfluidic organ-on-a-chip frameworks that precisely control fluid parameters and simulate reflux-like pathological processes; and exploring intraluminal perfusion-based exposure methods while preserving the three-dimensional architecture.
The esophageal chip developed by Palamà et al. based on the MIVO® platform provides an important paradigm. This model exposes the reconstituted epithelium to acid stimulation under dynamic fluid flow. It integrates multiple readouts—including transepithelial electrical resistance monitoring, permeability assays, quantification of inflammatory factor release, and histological analysis—to systematically evaluate mucosal damage and repair. This establishes a controllable and quantifiable analytical framework for assessing mucosal protectants. However, this model is currently based on immortalized cell lines. Although it offers significant advantages in dynamic control and multiparametric readouts, it still lags behind organoids in terms of cellular lineage diversity. 141 Consequently, future integration should focus on combining the engineered control of chip systems with the organoids.
Clinical Translation Barriers
The aforementioned limitations collectively constrain the progress of esophageal organoids toward clinical translation. As of January 2026, no personalized cancer treatment regimen based on this technology has received formal approval from major regulatory agencies, such as the U.S. Food and Drug Administration (FDA) or the European Medicines Agency (EMA), for routine clinical use. This underscores that the field remains in a critical phase of validation and dissemination.
The success of organoid construction depends heavily on the amount, viability, and overall quality of the initial biopsy specimen. Current culture systems fail to achieve the efficiency and stability necessary for clinical application. For instance, in ESCC, the initial success rate for establishing organoids is 60%, but only 10% of these models can be stably passaged beyond five generations (P5). 51 This instability compromises experimental reproducibility and the reliability of organoids as tools for precision decision-making. The lack of validation through large-scale prospective clinical studies limits the reliability and clinical applicability of these models. 142 Additionally, the absence of standardized protocols across laboratories hinders cross-institutional comparison and validation. Moreover, dependence on expensive reagents and specialized technical expertise increases costs, posing a financial burden on patients.
Regarding predictive performance, owing to inadequate tumor microenvironment recapitulation, drug sensitivity testing solely relies on tumor cell viability alterations, thereby limiting its clinical translational value. This reliance diminishes the reliability of extrapolating these results to clinical efficacy. Although some studies report high predictive accuracy, variations in endpoint definitions, drug exposure schemes, and response quantification hinder comparability across studies. More importantly, few large-scale, multicenter, prospective randomized controlled trials have used hard clinical endpoints, such as overall survival or progression-free survival, to provide primary evidence demonstrating that organoid-guided treatment strategies outperform standard therapy. Therefore, whether in vitro drug sensitivity data can be translated into tangible clinical benefit requires further confirmation through large cohort studies and clinical practice validation.
In terms of clinical application, there is a mismatch in timelines between organoid culture and clinical treatment decisions. Shen et al. reported a median time of about 3 weeks from patient sampling to the availability of a drug sensitivity report, ranging from 2 to 8 weeks. This period is theoretically shorter than the typical window period (about 2–3 months) for evaluating and adjusting treatment following therapy initiation. 130 This suggests potential for using the results from organoids in clinical practice. However, the adaptability of this technology in specific clinical scenarios remains limited. For patients requiring urgent intervention, such as those with distant metastasis and severe dysphagia, clinical guidelines recommend initiating treatment within days to few weeks. A testing duration of 2–8 weeks may surpass the recommended window for initiating first-line therapy. Consequently, organoid technology is suited for optimizing second-line therapy or managing recurrence. Moreover, current models are predominantly derived from surgical specimens of operable patients, underrepresenting those with advanced, unresectable, or frail conditions. This underrepresentation limits the universality of the models. The lack of dynamic tracking with paired pre- and post-treatment samples from the same patient makes it difficult to reveal the evolution of drug sensitivity and resistance. This constraint reduces the value of organoids throughout the course of cancer management.
To facilitate clinical translation and enable timely feedback, organoid systems can be integrated with high-throughput automated platforms to generate drug sensitivity data within clinically relevant timeframes. Jun et al. developed a high-throughput organoid-on-pillar (high-TOP) system that integrates automated organoid spotting with 384-well micropillar plates, overcoming challenges in standardization, throughput, and reproducibility in traditional 3D organoid screening. This system maintains temperature-sensitive extracellular matrices in a liquefied state during spotting, enabling precise deposition of cell–ECM mixtures onto predefined micropillars. Its 384-micropillar design supports highly parallelized 3D cultures, allowing easy media exchange, drug treatment, and staining without disturbing organoid domes. The platform also demonstrates high reproducibility, with a coefficient of variation (CV) of 6%–7%, thanks to automated non-contact dispensing and consistent micropillar geometry. Collectively, these features establish high-TOP as a reliable and standardized platform for ex vivo drug testing and precision oncology. 143
In conclusion, esophageal organoids can be translated from research tools into broadly applicable precision theranostic platforms only by overcoming current limitations—including insufficient standardization, genomic instability, incomplete recapitulation of the tumor microenvironment, and restricted dynamic functionality—and by optimizing application strategies tailored to different clinical contexts.
Future prospects
Esophageal organoids have become indispensable tools for advancing research and therapy in esophageal diseases. By closely recapitulating the structural and functional complexity of human tissues, these models offer substantial potential for disease modeling, drug screening, and personalized medicine. Continued methodological innovation is expected to address current limitations and enable the development of more physiologically relevant human models. This section outlines future directions for esophageal organoid research, focusing on the establishment of biobanks and shared resources, the integration of organoid intelligence, the construction of full-thickness models, immune co-culture systems, organ-on-a-chip technologies, and multi-organ interconnection platforms.
Esophageal organoid biobanks
Large-scale and standardized esophageal organoid biobanks will form the foundation for future biomedical research and clinical translation. At present, most esophageal organoid repositories consist primarily of PDOs from ESCC and EAC, with reported establishment success rates of approximately 60–70%.13,44 These PDOs preserve key features of the original tumors, including intratumoral heterogeneity, genetic alterations, and evolutionary trajectories, making them valuable tools for drug screening and resistance studies. However, existing biobanks remain limited in scale, are often restricted to single institutions, and lack standardized mechanisms for data sharing and quality control.
Future development should focus on several key areas. First, comprehensive and high-quality sample repositories should be established to cover the full disease spectrum, ranging from precancerous lesions—such as Barrett’s esophagus and squamous intraepithelial neoplasia—to invasive and metastatic cancers. These repositories should include matched normal tissues and adequately represent clinical heterogeneity across disease stages, histological subtypes, risk factors, and treatment histories. Second, organoid biobanks should be supported by multidimensional data systems. Each organoid line should be linked to complete clinical annotations and multi-omics datasets, including genomic, transcriptomic, epigenomic, and proteomic profiles. Standardized workflows, STR authentication, and routine quality control procedures will be essential to ensure reproducibility and data reliability.
In addition, future biobanks should evolve toward functionally integrated platforms. High-throughput drug screening, immune cell co-culture, and patient-derived organoid xenograft (PDOX) models can be incorporated to form a dual-track system that combines a “living sample repository” with a complementary “digital database.” Building on this infrastructure, an organoid–patient outcome linkage system can be developed to connect functional omics data with clinical responses. With appropriate ethical governance and data-sharing frameworks, such platforms would enable global access to high-quality esophageal organoid resources and accelerate the implementation of precision medicine in esophageal cancer.
Organoid intelligence
Organoid intelligence represents a paradigm shift in disease modeling and personalized medicine by integrating artificial intelligence (AI) with highly biomimetic organoid systems. In this framework, AI functions as the analytical engine, while organoids serve as biological carriers that faithfully recapitulate human tissue architecture. Standardized culture conditions combined with automated platforms enable closed-loop systems for organoid maintenance, monitoring, and quality assessment. Automated liquid-handling technologies reduce batch-to-batch variability and minimize human-induced error during organoid culture. 144
Recent advances in image-based analysis further enhance the capabilities of organoid intelligence. Deep learning platforms such as OrganoID 145 and deepOrganoid 146 employ convolutional neural networks to enable real-time quantification of organoid morphology, growth kinetics, and cell viability. Within this context, esophageal organoids offer unique advantages. ESCC and EAC display distinct morphological features, such as compact spheroids and glandular or cystic structures, which can be automatically classified and quantified using deep learning approaches. In Barrett’s esophagus models, longitudinal image analysis may facilitate early prediction of malignant transformation.
Machine learning models that integrate organoid pharmacogenomic data have already demonstrated success in predicting chemotherapy responses in colorectal and bladder cancers. Similar strategies can be extended to esophageal cancer by combining drug sensitivity profiles with key mutational data, including alterations in TP53, CDKN2A, and PIK3CA. 147 Such integration would enable the construction of personalized efficacy prediction models. Although challenges remain in algorithm generalization, clinical validation, and ethical governance, organoid intelligence is expected to drive continued convergence between biological simulation and computational prediction.
Full-thickness esophageal organoids
Current esophageal organoid models primarily recapitulate epithelial structures, whereas future efforts aim to generate full-thickness esophageal organoids incorporating the mucosa, submucosa, muscularis, and serosa. Recent studies in intestinal organoids suggest that replacing epidermal growth factor (EGF) with epiregulin (EREG) can promote the simultaneous differentiation of multiple lineages, including epithelial, mesenchymal, glial, and smooth muscle cells, within a single culture system. These models have demonstrated the formation of functional neuromuscular units capable of rhythmic contraction and neurotransmitter responsiveness. 148
Applying similar strategies to esophageal organoids may enable coordinated differentiation of the muscularis layer and intrinsic neural plexus. By adopting a “single-factor replacement–induced multilineage co-differentiation” approach and systematically screening niche factors involved in esophageal development, researchers may reconstruct full-thickness esophageal organoids with peristaltic function. Such models would facilitate mechanistic studies of esophageal motility disorders, including achalasia and diffuse esophageal spasm, and provide advanced platforms for investigating tumor invasion, metastasis, and radiation-induced esophagitis repair.
However, as organoid size increases, limitations in oxygen and nutrient diffusion become increasingly pronounced. To overcome these constraints, engineered vascularized support systems will be required. Integration with microfluidic and vascularized chip technologies may ultimately enable the generation of structurally intact, functionally robust esophageal organoids capable of long-term culture.
Immune co-culture systems
Immune checkpoint inhibitors have become standard treatments for advanced esophageal cancer, yet accurately predicting therapeutic efficacy and elucidating resistance mechanisms remain challenging. Current immune–organoid co-culture systems are relatively rudimentary and can be broadly categorized into two technical approaches. The first approach preserves endogenous immunity by retaining native immune cells and their spatial organization within tumor tissues. For example, the air–liquid interface (ALI) culture system developed by Neal et al. embeds fresh tumor fragments in a collagen matrix exposed to air, thereby preserving tumor-infiltrating lymphocytes and the native T cell receptor repertoire. This system has successfully recapitulated responses to PD-1/PD-L1 blockade in renal cell carcinoma, melanoma, and non-small cell lung cancer, and may be extended to esophageal cancer for short-term therapeutic assessment. 149
Despite its advantages, the ALI system is limited by the short in vitro lifespan of endogenous immune cells and its restricted applicability to small biopsy specimens. The second approach involves exogenous immune reconstruction, in which immune microenvironments are artificially reconstituted by co-culturing organoids with immune cells derived from peripheral blood. The gel–liquid interface (GLI) model established by Li et al. simulates systemic antitumor immunity by allowing peripheral blood mononuclear cells to migrate freely and infiltrate tumor organoids. This model has demonstrated strong concordance with clinical responses to PD-1 inhibitors in lung cancer patients. 150
Given that approximately 77% of ESCC cases exhibit immune-rejected 151 or immune-desert phenotypes and that EAC tumors are predominantly immune-depleted, 152 exogenous immune reconstruction strategies may be particularly important for esophageal cancer. Future immune organoid platforms should move beyond simple co-culture and toward multidimensional integration of immune, stromal, and vascular components to more accurately simulate dynamic tumor–immune interactions.
Esophagus-on-a-chip
Vascularized organ-on-a-chip technologies have been successfully applied to multiple organoid systems and offer transferable engineering frameworks for advancing esophageal organoid models. In vivo, the avascular stratified squamous epithelium of the esophageal mucosa relies on nutrient and oxygen diffusion from the vascularized lamina propria. When organoid size exceeds effective diffusion limits, central hypoxia and necrosis frequently occur, particularly in esophageal tumor organoids. Moreover, angiogenesis plays a critical role in esophageal cancer invasion and metastasis, which traditional avascular organoid models fail to adequately recapitulate.
Microfluidic platforms, such as those developed by Quintard et al., enable the formation of perfusable endothelial networks that anatomically connect with organoids and significantly enhance their maturation and functional performance. These systems provide a technical foundation for developing perfusable esophageal organoid-on-a-chip models. 153 In parallel, the combination of indirect three-dimensional bioprinting with microfluidic perfusion offers a versatile strategy for constructing vascularized organoids that integrate epithelial, stromal, and vascular components. By printing layered GelMA hydrogel structures and generating vascular channels using sacrificial Pluronic F127 templates, followed by endothelialization and perfusion, researchers can establish perfusable microenvironments with functional endothelial interfaces. 154
Given the layered organization of esophageal epithelium and lamina propria vasculature, such strategies provide a methodological basis for vascularized esophageal organoid chips. These platforms can support long-term culture of large organoids and enable studies of angiogenesis, metastasis, and anti-angiogenic drug responses, as well as applications in tissue engineering.
Multi-organ interconnection systems
The esophagus is functionally interconnected with multiple organ systems, including the stomach, pharynx, liver, and immune system. Future advances are likely to integrate esophageal organoids into multi-organ “human-on-a-chip” ecosystems. For example, microfluidic linkage of esophageal, gastric, and intestinal organoids could enable in vitro modeling of the full pathological continuum of gastroesophageal reflux disease, from acid and bile injury to Barrett’s metaplasia and adenocarcinoma development.
In addition, interconnected networks comprising esophageal, hepatic, and immune organoids can simulate first-pass metabolism and systemic immune responses, thereby enhancing the clinical predictive value of drug efficacy and toxicity assessments. From a systems biology perspective, such integrations elevate esophageal organoids from isolated tissue models to miniature representations of human physiology. Automated platforms, such as the “Interrogator,” have already demonstrated programmable fluidic connections and long-term maintenance of multi-organ chips while allowing dynamic reconfiguration of organ connectivity.
By establishing direct lumen-to-lumen pathways between esophageal and gastric organoids and applying controlled reverse perfusion of gastric contents, these systems can recapitulate key pathological stimuli of gastroesophageal reflux in vitro. Such highly biomimetic platforms will provide powerful tools for investigating reflux-associated esophageal diseases. 155
Although challenges remain in the modeling fidelity and clinical translation of esophageal organoids, their future developmental trajectory is increasingly clear. Progress will involve transitioning from static structural models to dynamic functional systems and ultimately to integrated, multi-organ platforms. Key priorities include the construction of full-thickness and immune-competent organoids, the application of engineered chip technologies to overcome microenvironmental and diffusion limitations, the establishment of standardized biological resources and intelligent analytical frameworks, and the realization of multi-organ interconnection. Together, these advances will position esophageal organoids as central tools in translational research and precision medicine.
In conclusion, the future of esophageal organoid technology holds great promise for advancing our understanding of esophageal diseases and revolutionizing clinical treatment. The path forward will be marked by innovative engineering approaches that integrate immune components, vascular networks, and neuromuscular function into more physiologically relevant models. As we continue to standardize protocols, expand organoid biobanks, and develop multi-organ platforms, the next-generation esophageal organoids will become increasingly powerful tools in personalized medicine and drug development.
Conclusion
The development and application of esophageal organoids have become pivotal platforms in disease modeling, precision medicine, and regenerative therapy. These highly biomimetic three-dimensional systems successfully recapitulate the cellular composition, structural complexity, and functional properties of native esophageal tissue. They offer transformative potential for understanding esophageal pathophysiology, screening novel therapeutic agents, and tailoring personalized treatment strategies. Esophageal organoids fill critical gaps in traditional models, offering unique insights into epithelial regeneration, tumor biology, inflammatory mechanisms, and microenvironmental dynamics. Despite remarkable progress, key challenges remain, including cultivation standardization, genomic stability, microenvironmental reconstruction, functional integration, and clinical translation. Methodological innovations, such as immune co-culture platforms, organ-on-a-chip technologies, full-layer model development, and multi-organ interconnection systems, are expected to overcome these limitations and expand the clinical applicability of organoids. Through interdisciplinary collaboration and technological optimization, esophageal organoid systems will continue to evolve as indispensable tools for advancing esophageal science, providing new approaches for unmet medical needs, and accelerating implementation of precision medicine.
Footnotes
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Henan Province Science and Technology Project (242102311164 and 252102310373).
Declaration of conflicting interests
The authors declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.