
Abstract
Digestive system tumors are the leading cause of cancer-related deaths worldwide. Despite ongoing research, our understanding of their mechanisms and treatment remain inadequate. One promising tool for clinical applications is the use of gastrointestinal tract tumor organoids, which serve as an important in vitro model. Tumor organoids exhibit a genotype similar to the patient’s tumor and effectively mimic various biological processes, including tissue renewal, stem cell, and ecological niche functions, and tissue response to drugs, mutations, or injury. As such, they are valuable for drug screening, developing novel drugs, assessing patient outcomes, and supporting immunotherapy. In addition, innovative materials and techniques can be used to optimize tumor organoid culture systems. Several applications of digestive system tumor organoids have been described and have shown promising results in related aspects. In this review, we discuss the current progress, limitations, and prospects of this model for digestive system tumors.

Introduction
Digestive system tumors primarily include esophageal cancer (EC), gastric cancer (GC), colorectal cancer (CRC), pancreatic cancer (PC), hepatocellular carcinoma (HCC), and biliary tract cancer, which are the leading causes of cancer-related deaths worldwide and have the highest incidence and mortality rates among malignant tumors. 1 In 2020 alone, nearly six million new cases of digestive system tumors were reported. 2 Currently, the primary treatment for early- and intermediate-stage digestive system cancers is surgery and postoperative adjuvant radiotherapy. Chemotherapy is the most common cancer treatment strategy; however, these drugs often suffer drawbacks, such as poor water solubility, low tumor targeting ability, and serious adverse effects, which greatly limit their clinical application. 3 Current in vitro cancer research models predominantly rely on 2D models, including tissue section culture and 2D cell line culture. 4 Tissue section models can capture transient interactions between physiologically relevant tissues, although they are often difficult to maintain for long periods and can rapidly lose their phenotype. 5 Currently, the most widely used model for tumor research is the patient-derived xenograft (PDX) animal model. Although the PDX model maintains the heterogeneity of the patient’s primary tumor, 6 the implantation rate of PDX models is limited, their application in personalized medicine is time-consuming, and they may disrupt the tumor microenvironment (TME).7–9
Compared with the PDX model, organoids require less time and cost to establish10,11 and only require the collection of a small piece of tissue from the patient biopsy, thus causing minimal harm to the patient. 12 Organoids are derived from pluripotent tissue progenitors (adult stem cells) and cancer cells, and are composed of multiple cell types with significant self-renewal and self-organization capabilities, maintaining the key structural and functional properties of organs.13,14 Compared to other models, organoids are more representative of typical physiological conditions. 15 Successful establishment of organoids for various digestive system tumors, including stomach, 16 colorectal, 17 liver, 18 pancreas, 19 gastroenteropancreatic neuroendocrine (GEP-NEN), 20 esophagus, 21 and bile duct 22 tumors, have been achieved. The development of gene-editing techniques and the successful establishment of organoids has important implications for the study of solid tumors.
This review discusses the current state of digestive tract tumor-related diseases and their treatment strategies, as well as the origins of digestive tract tumor organoids, and their characteristics, including their ability to retain the parent-related genotype and simulate the TME. We also highlight the lower cost, shorter time requirement, and higher success rate associated with organoid establishment. We then explore culture systems and the use of new materials and techniques for culturing digestive tract tumor organoids. Furthermore, we provide insights into GC, CRC, EC, pancreatic cancer, liver cancer, and gastrointestinal-pancreatic neuroendocrine tumor organoids. Finally, we conclude by discussing the various research applications of digestive tract tumor organoids as well as the current limitations and future directions for digestive tract tumor research.
Overview and characteristics of digestive tract tumor organoids
Overview of digestive tract tumors
Cancer is a disease caused by the loss of normal regulation and excessive proliferation of cells. In 2020, gastrointestinal cancers accounted for approximately 50.8% of all cancer cases worldwide. 23 Lifestyle factors, including obesity, excessive alcohol consumption, smoking, physical inactivity, and high cholesterol, are closely associated with the development of these cancers. 23 Among gastrointestinal tumors, GC and CRC have the highest incidence rates, 24 with an average 5-year overall survival of ~60%; however, survival rates decrease significantly after the onset of metastasis. 25 Symptoms of early GC are typically absent or mild and include nausea and vomiting. Patients with advanced GC present with more noticeable symptoms, including upper abdominal discomfort, satiety, aggravated abdominal pain, decreased appetite, and fatigue. 26 Similarly, early CRC is often asymptomatic or may cause mild discomfort, indigestion, or occult blood in the stool. As the tumor progresses, symptoms gradually worsen and include changes in bowel habits, abdominal pain, hematochezia, abdominal mass, ileus, anemia, fever, and weight loss. 27 The standard treatments for CRC are surgery, chemotherapy, and radiotherapy, which are often combined depending on the disease localization and progression.28–30 Multidisciplinary therapy is also crucial for effectively treating GC, cholangiocarcinoma, and HCC, utilizing systemic chemotherapy, radiotherapy, surgery, immunotherapy, and targeted therapy.31–33
Improved in vitro models are needed to gain a comprehensive understanding of tumor development and improve patient survival. Preclinical model systems are critical in guiding the development of targeted therapies by capturing inter- and intra-tumoral heterogeneity. Animal cancer models, especially genetically engineered mouse models, have provided valuable insights into the cellular and genetic basis of cancers. 34 However, these models are expensive, time-consuming, and not always clinically applicable due to differences between human cancer pathology and cell cultures. 35 In contrast, PDX models offer the advantage of retaining tumor characteristics and reflecting inter-tumor heterogeneity. 6 However, challenges persist, including the potential replacement of human tumor stromal cells and extracellular matrix (ECM) when transplanted into immunodeficient mice, which can disrupt the TME. 36 Therefore, a need exists for improved in vitro and in vivo models that better mimic human tumor characteristics, with a high success rate, low cost, and suitability for genetic modification, large-scale drug screening, personalized medicine, and other applications.4,37
Digestive tract tumor organoid profile
Intestinal organoids were first designed by Sato et al. 38 who established a specialized culture system using mouse crypts embedded in laminin-rich Matrigel, supplemented with different growth factors, including epidermal growth factor (EGF), Noggin, Wnt, and R-spondin. This system successfully established 3D mouse crypt structures resembling freshly isolated intestinal crypts, maintaining their characteristics for over 8 months. However, stem cells can improve the formation of organoid structures when co-cultured with Paneth cells. 39 More recently, organoids have gradually improved and become widely utilized in tumor research, especially in digestive system cancers, including CRC,40,41 GC,42,43 PC, 19 liver cancer, 44 gastroesophageal cancer,45–47 GEP-NENs, 20 pancreatic ductal adenocarcinoma (PDAC), 48 and EC 49 (Figure 1).

Timeline of digestive system tumor organoid development.
With continuous technological advancements, researchers have found that tumor organoids retain the heterogeneity of tumors between and within tissues, including gene expression profiles. This feature has the potential to advance tumor research and contribute to personalized medicine. 50 For example, in the case of GC, chemotherapy remains the main treatment method, however, drug resistance limits the clinical application of 5-FU. Zhou et al. 51 found improved therapeutic efficacy by the addition of STAT 3 inhibitors and simultaneously decreased resistance to immune checkpoint blockade immunotherapy. Therefore, exploring iron-mediated cell death triggered by STAT3 inhibition through organoids could serve as a therapeutic strategy against GC and chemotherapy resistance. 52 Regarding CRC, CRISPR-Cas 9 gene-editing technology has been employed to mutate tumor suppressor genes (APC, SMAD4, TP53, KRAS, and PI3K) in normal epithelial organs. Through the selective culture of colorectal organoids, CRC development can be effectively reproduced in vitro. This enables improved disease progression and drug screening analysis as well as individualized treatment for CRC. 53
In liver cancer, hepatitis B virus (HBV infection is the most prominent risk factor for HCC development. Currently, the models used in HBV research can be categorized into three types: cell models, animal models, and organoid models. While cell and animal models exhibit certain limitations in studying HBV-related carcinogenesis, liver organoids can be infected by recombinant virus and HBV, and key viral components, such as HBsAg, HBeAg, HBV core protein, and covalent closed circular DNA (cccDNA), can be detected in the culture supernatants. Accordingly, this model can help clarify the role of HBV in liver tumor pathogenesis. 54
Patient-derived organoids (PDOs) have been used to simulate in vitro tumor responses to drug treatment, the function of this model to screen drugs for guiding clinical decisions in cancer treatment is important. For example, Helen et al. 55 confirmed the feasibility of cancer organoids for drug screening by screening 37 anticancer drugs from nine GC organoids in seven patients. Thus tumor organoids provide a new approach for drug screening.
Characteristics of digestive tract tumor organoids
Simulating the TME
The TME refers to the microenvironment surrounding tumor cells and includes the surrounding blood vessels, immune cells, fibroblasts, bone marrow-derived inflammatory cells, various signaling molecules, and the ECM. If we consider tumor cells as seeds, the microenvironment they inhabit can be likened to the soil, with tumor cells and their microenvironment mutually influencing each other. 56 Regarding digestive system tumors, common clinical treatment options include surgery and postoperative adjuvant chemoradiotherapy. 3 Recently, immunotherapy has gained preference as it can indirectly alter the TME by supporting anti-tumor immune responses, improving the effectiveness of self-effector T cells and antigen-presenting cells (APCs), and inhibiting tolerant cells. Immunotherapy offers advantages, including low side effects, compared to other treatment modalities. 57 As we enter the era of immunotherapy, therapies targeting the TME are becoming increasingly diverse and complex. However, there is a significant gap in models that can accurately mimic the various immune components in the TME outside the human body. Despite recent advances in xenograft or humanized mice models, it remains challenging to observe heterogeneous anti-tumor immune responses in clinical trials of immunological oncology. Therefore, continued development of a more predictive model for assessing the effectiveness of clinical immunotherapies is needed. 58
Immune checkpoint blockade therapy has emerged as a prominent approach in cancer immunotherapy. By blocking immune checkpoints, this therapy activates endogenous anti-tumor T cells, effectively breaking down the body’s natural barriers and eliminating cancer cells. 59 Over the years, significant progress has been made in studying and targeting two important immune checkpoint pathways: PD-1/PD-L1 and CTLA-4/B7. 60 For example, Li et al. 61 demonstrated the feasibility of anti-PD-1 drugs for the treatment of HCC. However, tumor organoids cultured solely from tumor epithelial cells cannot effectively retain the complex diversity and physical structure of the TME. Specifically, these organoids lack the ability to mimic immune checkpoint blockade due to the absence of naturally infiltrating immune cells. 62 Recent advances in PDOs have addressed this limitation. That is, PDOs derived from over 100 human biopsies or mouse tumors, in homologous immune-competent hosts, have been successfully cultured at the air-liquid interface (ALI). This facilitates the incorporation of naturally embedded immune cells (T, B, natural killer [NK], and macrophages) within the tumor epithelial spread. Consequently, human and mouse PDOs have demonstrated the ability to mimic immune checkpoint blockade. These organoids exhibit functional activation of the PD-1/PD-L1 checkpoint blockade, enhanced cytotoxic responses, and the retention of various endogenous immune cell types, including macrophages, B cells, and NK cells, over a 7-day assessment period. 62 Notably, progress has also been made in the field of digestive system tumor organoids. For example, Tsai et al. 63 co-cultured native organoids with patient-matched peripheral blood lymphocytes and cancer-associated fibroblasts (CAFs), enabling the study of interactions between immunotherapy and tumor immune cells. Additionally, studies with pancreatic cancer organoids have provided insights into the activation of myofibroblast-like CAFs and the infiltration of the ECM in tumor tissues. These models have also proven valuable for assessing T cell infiltration and the activation of cytotoxic lymphocytes influenced by immune checkpoint blockade. In CRC, co-culturing autologous tumor organoids with peripheral blood lymphocytes has facilitated the enrichment of tumor-reactive T cells from peripheral blood of mismatch repair-deficient CRC, thereby enhancing our understanding of the pathways involved in tumor cell susceptibility and resistance to immunotherapy. 64
Although organoids exhibit great potential as preclinical models for immunotherapy, they may differ from the actual TME due to the potential immunogenicity of cells and batch differences in the Matrigel used for culture. 65 To this end, with the continuous development and improvement of technology, tumor organoids have the potential to replicate in vivo tumors, including their heterogeneity, TME, and gene expression. This will greatly contribute to the research and development of cancer drugs and drug screening.
Genetic similarities with parental-origin genotypes
A significant understanding of cancer has been achieved through genome analysis and expression profiling of clinical specimens. However, tumors comprise various cell types, including malignant cells, immune cells, and stromal subgroups. Thus, to study tumor composition and functional heterogeneity, single-cell transcriptome sequencing technology (scRNA-seq) is often employed. 66
In the case of digestive tract tumors, tumor organoids with partial gene expression profiles have been established. Wang et al. 67 dissociated tumors from patients with colon cancer into a single-cell suspension and performed single-cell RNA-seq analysis on half of the cells, while the other half was used for tumor organoid culture. They found that tumor tissue or tumor-derived organoids exhibited a higher percentage of stem-like cells. Differential gene expression analysis was then conducted on tumor and normal tissues, revealing that the colon cancer organoids exhibited high expression of tumor-specific genes associated with disease progression and metastasis (e.g. PROCR, SCD, BMP4, CEACAM6, TESC, and TGFBI). Similarly, Miyabayashi et al. 68 proposed a new xenograft model (IGO) for human pancreatic ductal adenocarcinoma. By intraductal injection of isolated organ tissue, this model distinguished between fast and slow human pancreatic ductal cancer progressors based on the bimodal distribution of survival rates. Global mRNA sequencing of the IGO model identified genes related to EMT, proliferation, and cell cycle (i.e. MYC targets, E2F targets, and G2M checkpoint signatures) enriched in rapid progressors. Genes associated with PDAC progression, such as KRAS and mTORC1 signaling genes, were also enriched in rapid progressors. Indeed, rapid progressors showed clear enrichment of squamous PDAC and basal-like features, while slow progressors exhibited enrichment in genes related to bile acid metabolism and fatty acid metabolism signaling. Slow progressors also displayed significant enrichment of ancestral-type PDAC and classical features. Thus, the distribution of molecular subtypes in the IGO model reflects the relative distribution of subtypes observed in human PDAC tumors, providing an opportunity to investigate the relationship between PDAC subtypes and human specimens.
Given the wide genetic mutation spectrum and lack of clear characterization in hepatobiliary tumors, Zhao et al. 69 generated scRNA-seq profiles using established patient-derived hepatobiliary tumor organoids, including HCC, intrahepatic cholangiocarcinoma (ICC), and gall bladder carcinoma. They identified common genes expressed in all three organoid types, including CTNNB1, HNRNPH1, and PPP1CB. These organoid tissues exhibited two distinct classes of genes: the widely recognized housekeeping gene, GAPDH and NEAT1. Additionally, genes associated with malignancy, including MET, PIK3R1, PRKCA, PTEN, SHC1, and STAT3, were specifically expressed in HCC organoids. This suggests genes similar to the parents in hepatobiliary tumor organoids. Notably, GAPDH promoted liver tumorigenesis through glycolysis. 70 Meanwhile, NEAT1, an important nuclear lncRNA and paraspeckle structural component, was implicated in HCC development under hypoxic conditions. 71 Hence, tumor organoids retain a portion of the genotypic characteristics of the parental tumor, making them a valuable resource for guiding clinical anti-cancer drug screening, personalized treatment, and precision medicine.
Comparison with the PDX model
The PDX model involves implanting patient tumor tissue directly into immunodeficient mice, allowing the establishment of tumor tissue in vivo. This model has been widely used in cancer research and in vitro cancer models 72 as they retain tumor heterogeneity and exhibit stability within the body. At the molecular level, PDX models retain the original phenotype and molecular characteristics of cancer. In contrast to tumor organoids, these models can also predict clinical outcomes, making them valuable for preclinical drug detection, personalized medication strategies, and precision medical treatment.73,74
In the case of digestive tract tumors, PDX models are widely used. For example, Misale et al. 75 used PDX from patients with CRC and quadruplex wild-type profiles (BRAF, KRAS, KRAS, NRAS, and PIK3CA) and concluded that the MEK inhibitor pimasertib alone does not effectively inhibit tumor growth, whereas the EGFR inhibitor cetuximab significantly reduces cancer proliferation by more than 70%. Similarly, van de Wetering et al. 76 detected resistance to anti-EGFR inhibitors cetuximab and BIBW2992 (afatinib) in KRAS mutant organoids. Thus, PDX models exhibit similar resistance detection capabilities as tumor organoids. Moreover, PDX models of patients with CRC retain the morphological and genomic features of the original tumors, exhibiting consistent mutation status for KRAS, NRAS, BRAF, and PI3K. 77 These mutations are also represented in CRC organoids. 76 However, PDX models have certain disadvantages. First, there is a time gap between obtaining tumor tissue from the patient and its clinical treatment, which delays the real-time application of personalized medicine. That is, PDX model preparation typically takes 4–8 months before conducting preclinical studies. Unfortunately, this timeframe is often not feasible for many patients. 78 In contrast, organoid models can be established within 4–12 weeks, allowing for rapid drug screening, and selection of the most suitable treatment regimen. 79
Second, intratumoral inhibition contributes to the accumulation of variability in PDX models during implantation and passage. This variability may increase with successive transplantations, leading to potential selection bias during the sampling of PDX models for implantation and transplantation. 36 Careful selection of tissue sections is necessary for the successful establishment of PDX models, ensuring that only the most suitable tissue containing essential elements is implanted. For example, pancreatic tumors are solid tumors, and proper classification of stromal and vascular cells is crucial to avoid adversely affecting experimental results. 80 In contrast, PDO is able to retain the tissue structure and tumor differentiation with high fidelity. 81
Culturing digestive tract tumor organoids
Traditional culture systems
In 2011, Sato et al. 40 pioneered the establishment of tumor organoids. Using the culture conditions originally developed for mouse colon crypts, they successfully generated organoids from the small intestine and colon by adjusting the culture environment. They further refined this approach to optimize the production of tumor organoids derived from colon adenoma, adenocarcinoma, and Barrett’s esophagus. Wetering et al. 76 applied a similar approach to generate 22 CRC organodis from 20 patients.
To culture different tumor organoids, tumor cells, including tumor stem cells, are isolated from patient tumor tissue and cultured in a 3D environment. By adding specific cytokines and small molecules, a 3D culture with a microenvironment resembling the human body is established, resulting in a miniature tumor model.62,82 The most commonly used matrix substrate, that is, Matrigel, is used to maintain the 3D culture. However, Matrigel, derived from the secretions of Engelbreth-Holm-Swarm mouse sarcoma cells, has an unclear composition and can introduce variability in experimental results. 83 Additionally, it is crucial to determine the composition of the induction medium, which involves adding various factors to the basal medium Dulbecco’s modified eagle medium (DMEM)/F-12. Considering that genetic mutations can vary among tumor tissues, the induction factors for the same tumor organoids may differ. Generally, growth and inhibitory factors, such as Wnt signaling pathway activators (Human-R-Pond-1) or inhibitors (SB202190), and transforming growth factor β (TGF-β) inhibitors (A83-01), are added to the culture medium. These factors are essential throughout the tumor organoid culture process.10,84,85 Consequently, there is room for improvement in selecting induction factors and culture methods to cultivate tumor organoid models that closely resemble the parent type.
Advancements in novel materials for organoid culture
Decellularized ECM and recombinant ECM proteins
Matrigel is commonly used in organoid culture due to its versatility and affordability. However, it is a complex substance with proteome analysis revealing 1800 unique proteins. 83 In the 3D culture system, the mechanical properties of Matrigel can greatly influence cell, organoid, tissue, and organ development. However, these mechanical properties are difficult to separate from the chemical cues provided by Matrigel. Moreover, Matrigel’s heterogeneous mechanical properties and potential immunogenicity pose challenges to its application in human clinical transplantation. 86 Given these limitations, there is an emerging need to develop Matrigel-independent organoid culture methods, including decellularized ECM, synthetic hydrogels, and gel-forming recombinant proteins (Figure 2(a)). 86
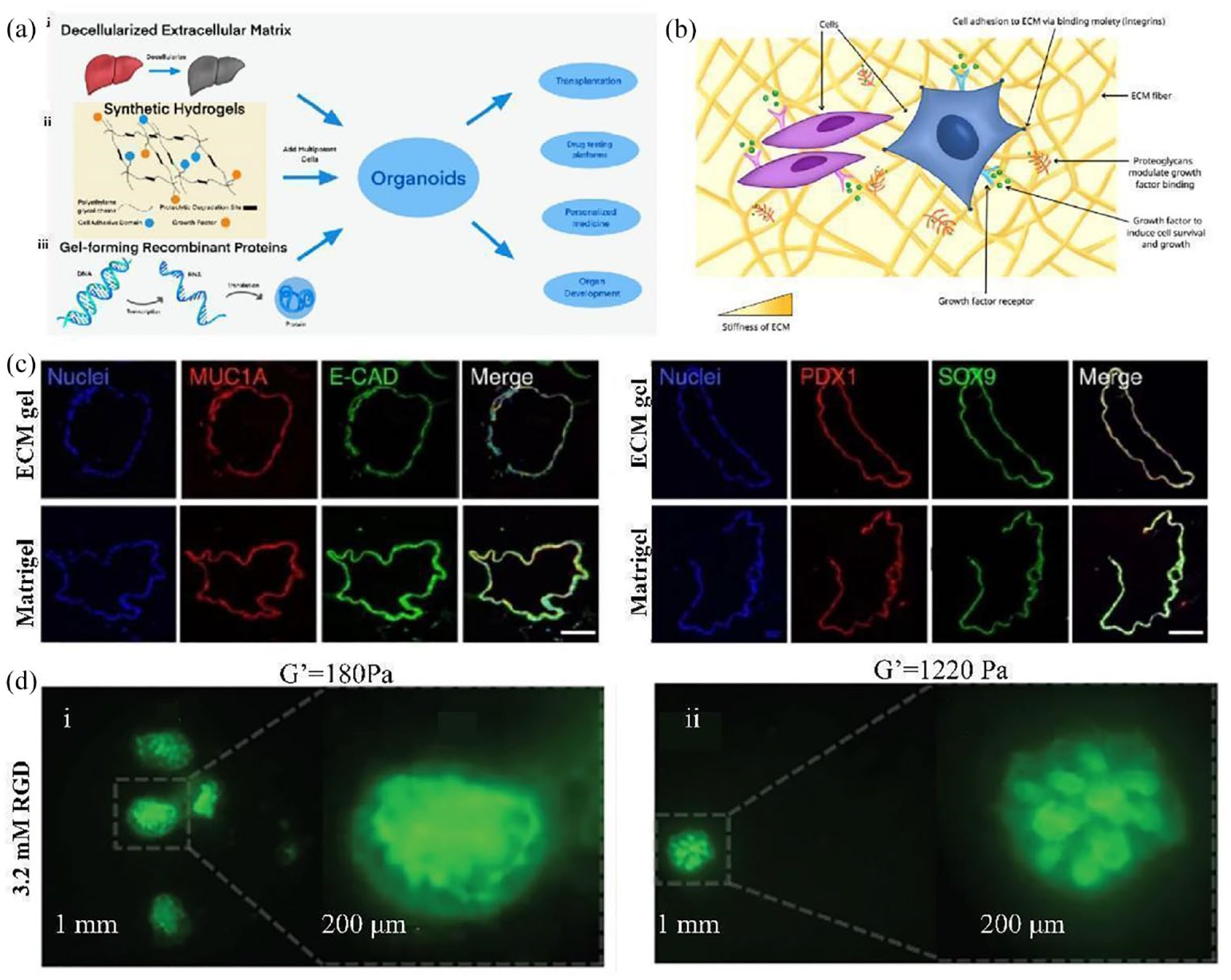
Methods of making organoids with decellularized ECM and recombinant ECM proteins. (a) Three kinds of organoid culture methods without Matrigel. (i) Decellularized extracellular matrix. (ii) Synthetic hydrogels. (iii) Gel-forming recombinant peptides. Adapted from Kozlowski et al. 86 (b) Microenvironment of cells. Adapted from Kozlowski et al. 86 (c) Some specific markers in acellular cultured pancreatic organoids. Adapted from Giobbe et al. 87 (D) Fluorescence images of intestinal organoids of GFP adult mice cultured in the eECM matrix for 3 days. (i) Organoids of shear moduli of 180 Pa from first day to third day. (ii) Organoids of shear moduli of 1220 Pa from first day to third day. Adapted from DiMarco et al. 88
During organoid culture, ECM proteins provide signaling cues, serving as adhesion substrates, and sequestering growth factors (Figure 2(b)). Decellularized ECM, in particular, provides additional signaling cues for tissue regeneration. This approach has been successfully demonstrated in gastrointestinal organoid culture. For example, Saheli et al. 89 demonstrated this approach in liver organoid culture by utilizing a sheep liver-derived ECM hydrogel (LEMgel) prepared through an optimized decellularization method. The LEMgel contained high levels of glycosaminoglycan, collagen, laminin, and fibronectin. By inoculating a combination of human HCC cells, mesenchymal stem cells, and umbilical cord stem cells into the LEMgel, they generated tumor organoids that exhibited superior functional activity compared to those cultured in type I collagen or anhydrous gels. The resulting tumor organoids demonstrated enhanced hepatocyte functionality. Meanwhile, Giobbe et al. 87 developed a method for decellularizing the pig small intestine to form an intestinal ECM gel that supports the cultivation of mouse Lgr5 crypt cells, as well as human pediatric gastric, intestinal, liver, gastric, and pancreatic organoids. The pancreatic organoids cultured by decellular cells maintained the expression of certain specific markers, including mucin-1a, epithelial cadherin, pancreatic-duodenal homeobox 1 (PDX1), and SOX9 (Figure 2(c)). 87 Additionally, naturally-derived proteins like type I collagen have been used to supplement decellularized ECM. For example, collagen I isolated from rabbit colon smooth muscle cells was used to culture human CRC model organoids. However, these tumor organoids exhibited decreased viability and were not sensitive to 5-fluorouracil chemotherapy treatment. 90 Therefore, the advantages of acellular ECM in tumor organoid culture, as well as the use of naturally-derived proteins, such as collagen I, require further investigation.
Besides acellular ECM, recombinant ECM proteins produced through biological genetic engineering have gained popularity due to their ability to incorporate accurately defined chemical cues. For example, recombinant ECM proteins have also been used in pancreatic organoid culture. Ghazalli et al. 91 developed an artificial elastin-like polypeptide called artificial aECM-lam, containing an 18 amino acid sequence from α1 laminin. This polypeptide was then combined with methyl cellulose in a medium and applied to pancreatic organoid culture in place of Matrigel. Interestingly, aECM-lam performed better than pancreatic organoids. The researchers compared the effects of aECM-lam, aECM-scr (containing interfering sequences instead of IKVAV), and Matrigel on single cells cultured in a semi-solid medium and found that the aECM-lam medium induced superior endocrine effects in the pancreas, while Matrigel inhibited the expression of endocrine genes. Matrigel also inhibited the development of endocrine and coniferous cells, while promoting ductal progenitor differentiation. 91 Furthermore, aECM-lam can generate adult progenitor cells from the mouse pancreas, which primarily express CD133 and low levels of CD71. 92
Recombinant proteins in organoids can be modified through protease recognition sites or chemical crosslinking to adjust stiffness, viscoelastic behavior, and chemical functionality. However, the diversity of proteins limits the feasibility of creating recombinant protein ECM, and certain proteins may elicit an immune response. Therefore, proteins expressed in mammals are typically selected to avoid unnecessary immunogenic factors. 86 Through a recombinant engineering strategy, DiMarco et al. 88 altered the cell adhesion binding domain concentration and matrix stiffness of the engineered extracellular matrix (eECM) independently to form a hydrogel with a shear modulus of 180 and 1220 Pa and an RGD concentration of 3.2 mM, successfully cultivating primary adult mouse intestinal organoids (Figure 2(d)). However, currently, there is limited research on the application of recombinant ECM proteins in tumor organoids, making it uncertain whether they would be more effective in this context. Further studies are needed to explore their potential benefits in tumor organoid cultures.
Alginate
In addition to Matrigel, acellular ECM, and recombinant ECM, alginate is also employed for organoid culture. Alginate is a linear anionic polymer isolated from marine brown algae. It can form alginate-based hydrogels via gentle and rapid gelation with divalent cations, such as Ca2+ and Ba2+. Alginate offers many advantages in biomedical applications, including stable and rapid gelation at room temperature, non-toxicity, and low cost, while avoiding the disadvantages associated with animal-derived products. 93 An important feature of alginate is its inherent cell adhesion properties and limited cell-to-cell interactions, which favor encapsulating cells (Figure 3(d)). 94 Alginate can also be used in cancer treatment (Figure 3(a)). 95 In addition, various materials can also be incorporated into the alginate to improve its printability or bioactivity and make it a suitable bioink(Figure 3(c)). 96 Due to the relative biological inertia of alginate, it is impossible to differentiate cells into organoids. However, other cells can be used to compensate for this defect. For example, Capeling et al. 97 found that non-adhesive alginate can provide mechanical support for human intestinal organoids (HIOs), and promote their growth by enveloping them with interstitial cells. Alginate has also been used to generate human pancreas, 98 and human intestine 97 organoids. Comparing organoids cultured in alginate to those cultured in Matrigel, HIO cultured in alginate exhibit similar developmental timelines, molecular structures, differentiation, and maturation, while being more cost-effective and easier to use. 97

Culture organoids with alginate. (a) Alginate-based material preparation process for cancer therapy. Adapted from Zhang et al. 95 (b) Schematic representation of RNA sequencing analysis of epithelial cells from HIO grown from alginate and matrix glue. Adapted from Capeling et al. 97 (c) 3D bioprinted ink were prepared by alginate, TOCNFs, and PDANPs. Adapted from Im et al. 96 (d) Encapsulation of cells with alginate. Adapted from Andersen et al. 94
In addition, combining seaweed-derived alginate with other techniques has yielded positive results in tumor organoid culture. For example, Fang et al. 99 demonstrated that by encapsulating tumor debris in non-viscous alginate and employing microfluidic droplet technology, tumor organoids with epithelial cells resembling the original tumor and maintaining the tumor phenotype can be produced. These organoids exhibit different responses to drugs like doxorubicin and ramicin A, making them suitable for drug screening. Additionally, the combination of alginate and bioprinting technology has demonstrated promise. Flores-Torres et al. 100 developed patient-derived biological tissue printing models using hydrogels composed of alginate, gelatin, and Matrigel. These models can be utilized for drug screening, maintaining and expanding patient-derived cancer spheroid cultures, and preserving cell activity and division rates. Moreover, Capeling et al. 97 used RNA sequencing analysis to determine the high molecular similarity of HIO epithelium both in vitro and in vivo (Figure 3(b)). Overall, alginate is a promising material for further exploration in the cultivation of tumor organoids.
Culturing organoids using novel techniques
Microfluidic technology
In recent years, progress has been made in the application of microfluidics in 3D cell culture. Schuster et al. 101 described a microfluidic organoid culture system that is compatible with gel systems and allows automated and high throughput (Figure 4(a)). This system enables real-time and reproducible analysis of organoids to advance the current understanding regarding their growth, morphology, and biochemical composition. In addition, droplet-based microfluidic approaches require a smaller sample size for multi-condition screening, and can control the degree of supersaturation (Figure 4(b)). 102 Therefore, this system can be used for high-throughput drug screening. Xu et al. 103 introduced a microfluidic chip design with an integrated 3D co-culture that enables drug susceptibility testing for anticancer drugs. They accurately replicated the TME system by culturing lung cancer tissue in 3D cultures. A gradient concentration generator (CGG) was later introduced, confirming the feasibility of the device for drug screening.

Application of microfluidic technology in cultivating tumor organoids. (a) Automated microfluidic 3D cellular and organoid culture platform for dynamical drug perturbations. (i) A programmable membrane valve-based micro fluidic chip. (ii) 3D culture chamber platform(scale bar 100 μm). (iii) Cross-section of a pdms-based bilayer multi-chamber 3D culture chamber device(scale bar 100 μm). (iv) Automated and dynamic drug screens. (v) Continuous observation of organoid or 3D cellular structures by time-lapse imaging. (vi) Quantification of organoid or 3D cellular structures. Adapted from Schuster et al. 101 (b) Formation of protein crystals in microfluidic droplets via the control of supersaturation. Adapted from Linsenmeier et al. 102 (c) Schematic showing the functionalization and regeneration process of the electrodes and image of the electrochemical sensor for biomarkers detection in an OOC platform. Adapted from Zhang et al. 104
The use of Matrigel for organoid culture can lead to differential microenvironments among organoids of the same species, limiting their ability to accurately mimic cancer-microenvironment interactions. To address this issue, the concept of organ chips has emerged. An organ chip is a functional unit that can imitate the organ-level physiology of the human body in vitro. It is constructed by replicating the anatomical structure of organs and assembling them in vitro with the necessary components for physiological function. 105 More specifically, organ chips are microfluidic cell culture devices typically made of polymers, such as polydimethylsiloxane (PDMS). They house living cells and can combine multiple cell types, while simulating the ability of perfusion blood vessels to provide metabolic requirements for tumor growth and replicate transport characteristics of the TME.106–108 Moreover, organ chips allow the integration of multiple microorgans in separate microcompartments connected by microfluidic channels, effectively forming a human microphysiological system (MPS). MPSs can simulate the structural and functional complexity of human tissues and organs. 79 Engineered patient tumor organoids integrated with MPSs have demonstrated the ability to mimic immune checkpoint blockade responses against programmed death 1 (PD-1) and programmed death ligand 1 (PD-L1) antibodies. They can also activate antigen-specific tumor-infiltrating lymphocytes and induce tumor cytotoxicity. By utilizing organ chips, these systems provide insights into the diverse alterations within the immune TME.109,110 Therefore, the organ chip technology can effectively solve the problem that the organoids are difficult to simulate the tumor microenvironment.
Certain tumor chip models, established using tumor cells, may not fully represent tumor properties. In contrast, tumor organoids maintain similarities with the primary tumor of the patient but lack precise control over the regulation of the immune TME. This raises the idea of whether the combination of organoids and organ chips can truly complement each other in the context of the TME. In pursuit of an answer, an organoid chip model has been proposed. 111 However, organoid chips continue to face challenges regarding fidelity and reproducibility. 112 For example, Kasendra et al. 113 combined an intestinal chip with organoids by culturing normal intestinal epithelial cells as organoids and implanting them onto a porous membrane of a microfluidic chip. Through transcriptomic analysis, they found that the organoid chip structure exhibited higher fidelity than organoids or organ chips alone. However, further verification is needed to determine if the obtained model can accurately maintain similarity to the patient’s tumor.
Microfluidics offers an improved approach for culturing organoids and facilitating their vascularization. Traditional organoid cultures often face limitations in nutrient supply due to restricted medium diffusion. Consequently, organoids rapidly achieve their maximum growth, leading to necrosis in the core as oxygen, nutrients, and metabolites fail to reach it through diffusion. This challenge can be overcome using organ chips. 114 In particular, gut-on-a-chip (GOC) systems can advance our understanding of intestinal physiology and the cause of disease. Moreover, Zhang et al. 104 implemented label-free biosensors using antibody-functionalized gold microelectrodes on a fully integrated modular heart-liver-chip multisensor platform for automated online monitoring (Figure 4(c)). Berger et al. 115 used microfluidic technology to transport nutrients, metabolites, and oxygen to organoids through laminar flow, preventing the formation of a “dead core” in smaller organoids. Microfluidics also enables the expansion of endogenous reservoirs of endothelial progenitor cells within organoids, inducing blood vessel formation. 116 In short, continuous advancements in microfluidic technology hold the potential to optimize organoid culture methods further.
3D bioprinting
A 3D bioprinting is an emerging technology that constructs artificial tissue or organ structures by designing the desired target and utilizing 3D printing methods combined with biomaterials. 117 It enables the creation of complex 3D structures in an automated manner and can be used to build the required tissues and organs. That is, 3D bioprinting produces the scaffolds with controlled physical properties, spatial heterogeneity, and the cell composition of ECM tissue, while accurately controlling the spatial distribution of cells and the ECM (Figure 5(a)). 118 For example, PDAC models printed in 3D can better mimic the natural structure of PDAC organization. 119 During this process, the desired artificial organs and scaffolds are formed by assigning small units of cells and biomaterials with micron-scale accuracy (Figure 5(b)). 120
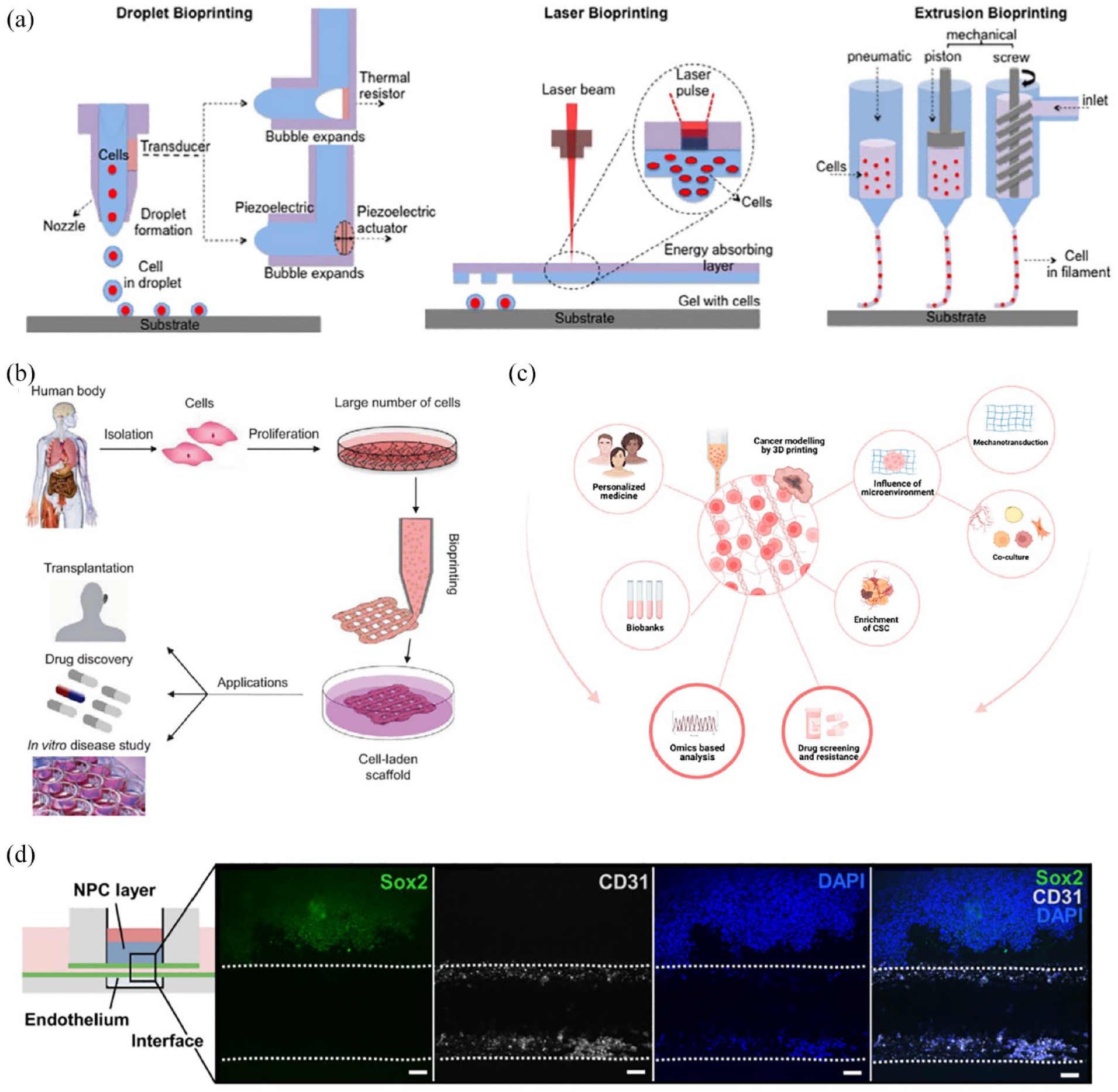
Tumor organoids based on 3D bioprinting. (a) Schematic diagram of 3D bioprinting approaches. Adapted from Arslan-Yildiz et al. 118 (b) Workflow for 3D bioprinting. Adapted from Mandrycky et al. 120 (c) Application of 3D bioprinting in cancer. Adapted from Datta et al. 121 (d) Bioprinted tissue-on-chip with vascular-like channel. Adapted from Duarte Campos et al. 122
Bioprinting technology comprises four main types: inkjet bioprinting, extrusion bioprinting, lithography, and spheroid bioprinting/bioassembly. 123 Each type offers distinct advantages. More specifically, lithography enables high-resolution features, between 5 and 10 µm, and complex geometries. Spherical biological printing allows for heterogeneous components with high spatial precision. Extrusion biological printing provides fast processing times and various commercial printers. Finally, inkjet offers high precision and low shear stress.123,124 Grix et al. 125 used stereo lithography to combine HepaRG and human stellate cells to produce bioprinted liver organoids that exhibit basic properties of the liver.
Bioink is a hydrogel prepolymer solution that encapsulates cells 120 and exhibits biocompatibility, printability, mechanical and structural integrity, biomimicry, and biodegradation. However, different bioinks have unique effects on cells during the bioprinting process. 126 For example, alginate-based hydrogels mixed with gelatin methacrylate (GelMA) enhance printability and cellular affinity, while the addition of four-arm poly(ethylene glycol)-tetraacrylate enhances the crosslinking density and mechanical strength. Moreover, decellularized ECM isolated from tissues or organs can act as a bioink to simulate the complex ECM microenvironment. Meanwhile, bioinks synthesized from methacrylic acid (ma) and polyethylene glycol (PEG) exhibit rapid polymerization and high mechanical stability. 127 Duarte Campos et al. 122 reported that hiPSC-derived neural progenitor cells, printed with ELP hydrogel and the fibronectin-derived, cell-adhesive RGD amino acid sequence (ELP-RGD) as the bioink, maintain their stem-like phenotype and express their specific marker Sox-2 (Figure 5(d)).
As our understanding of cancer improves and in vitro models become more abundant, we recognize the importance of the TME in tumor growth, progression, and drug resistance.121,128 Although tumor organoids can partially reconstruct the TME using the ALI method, fully accurate simulation is not achieved due to the absence of blood vessel formation and multiple cell types. However, 3D bioprinting allows precise control over the deposition of biological agents through various techniques, facilitating the establishment of vascular networks to accurately simulate the TME (Figure 5(c)). 121 Another study showed that bioprinted organoids retained the high-order structure of cancer cells. That is, by using pancreatic ductal adenocarcinoma patient-derived tumor tissue enzymes as the bioink, the printed organs accurately reflected the characteristics of cancer cells. Bioprinting also preserves the similarity to patient tumors and maintains the vitality and sensitivity of cancer cells to chemotherapy drugs. 129 Hou et al. 130 used magnetic bioprinting to produce pancreatic cancer organoids, which effectively promoted the generation of PDO models and improved the efficiency of drug screening.
Crispr-hot
The CRISPR-Cas system is a prokaryotic acquired immunity mechanism that enables bacteria to employ the CRISPR-Cas 9 tool for DNA cleavage, providing defense against viruses. This system precisely modifies the DNA of organisms, making it one of the most popular gene-editing techniques. 131 The CRISPR-Cas system operates through CRISPR sites composed of CRISPR arrays, which contain spacer sequences that act as genetic memory, conferring resistance to viruses with matching sequences. The arrays include a key Cas protein, responsible for cleaving the target DNA, leading to a double-strand break. This protein is an essential component of all CRISPR-Cas systems, as it is crucial for the adaptive immune response, which comprises three phases: adaptation/spacer acquisition, pro-crRNA expression, and interference. 132 Among the different CRISPR systems, Type I, which is prevalent in bacteria and archaea, is widely utilized for gene-editing. This system functions through the CRISPR-associated complex (Cascade), a multi-subunit effector complex, and crRNA to defend against viral threats. Cascade binds to the target DNA, forming the R-loop structure, and recruits Cas3 to cleave the target DNA. 133 Indeed, studies on the CRISPR-Cas mechanism will benefit the establishment of organoids (Figure 6(c)). 134

The application of CRISPR-Cas9 in tumor organoid culture. (a) Ex vivo strategy for gene therapy based on CRISPR/Cas 9. Adapted from Savić and Schwank. 135 (b) Driver gene validation was performed on mouse intestinal tumor organoids using CRISPR-Cas 9. Adapted from Takeda et al. 136 (c) The mechanism of CRISPR/Cas9. Adapted from Zhan et al. 134 (d) Genetic engineering of pre-tumorigenic organoid line (AK: APC-KO and KRASG12D) and a line that is tumorigenic after subcutaneous transplantation (AKT: additional TGFBR2-KO). Adapted from Michels et al. 137
Compared to other site-specific endonuclease technologies, such as zinc finger nuclease (ZFN) and transcriptional activator-like effector nuclease (TALENs), the CRISPR system offers an advantage by using single guide RNA (sgRNA) to target genome sequences, eliminating the need for protein-based detection methods, such as TALENs and ZFN. The CRISPR-Cas 9 technology can also be used for gene therapy in vitro by editing a patient’s stem or progenitor cells before transplanting them back into the patient, a method that may prove useful in resolving human immunodeficiency virus (HIV) infection (Figure 6(a)). 135
The CRISPR-Cas 9-mediated homologous independent organoid transgenic (CRISPR-HOT) approach allows for the efficient integration of exogenous genes into organoids, facilitating the development of various knock-in human organoids. For example, Artegiani et al. 138 compared the knock-in efficiency of the CRISPR-HOT method and homology-directed repair at the TUBB site in human hepatocyte organoids. They discovered the knock-in efficiency was higher using the CRISPR-HOT method, while both knock-in organoids exhibited identical fluorescent signals. Additionally, Hendrisks et al. 139 conducted genome engineering of a human fetal hepatocyte organoid culture via gene knock-in and knockout. Such studies provide insights into hepatocyte division, ploidy, and the role of genes in liver cancer. In conclusion, using the CRISPR-HOT approach can improve the efficiency of integrating foreign genes into organoids.
These advancements highlight how CRISPR-Cas 9-mediated genome editing can enhance the ability to accurately simulate cancer in organoids. For example, sequential mutations introduced by CRISPR-Cas9, targeting genes such as APC, TP 53, KRAS, and SMAD4 in intestinal organoids, can recapitulate the progression of CRC in vitro. 76 Moreover, Michels et al. 137 established a colon organoid engineered with loss of APC; the oncogenic KRASG12D allele (AK organoids) exhibited growth only after additional deletion of TGFBR2, providing a sensitive gene-based system (Figure 6(d)). AK organoids retain a genetically stable diploid phenotype in vitro and can be efficiently modified by CRISPR-Cas9. Meanwhile, Using CRISPR-Cas 9 to modify mouse intestinal tumor organoids and CRC organoids (Figure 6(b)), Takeda et al. 136 verified that Acvr1b, Acvr2a, and Arid 2 act as tumor suppressors in CRC.
In summary, CRISPR-Cas 9 technology holds great potential for advancing gene-editing in organoids and improving our understanding of various diseases, including cancer.
Classification of digestive tract tumor organoids
GC organoids
In 2020, the global incidence rate of GC was 5.6%, with a mortality rate of 7.7%. 2 Understanding the mechanism underlying GC development is crucial for improving treatment regimens and developing novel anti-cancer drugs. Organoid models offer a valuable approach to studying developmental mechanisms, as they accurately simulate biological processes and provide insights into tissue renewal and stem cell function.140,141 GC organoids were first established from the tumor biopsy tissue of patients (Figure 7(a)). 142 In the case of gastric organoid culture, Huch et al. 143 employed antral glands isolated from the stomach and embedded them into the ECM. Through this research, they identified stomach-specific niche factors, including WNT, FGF-10, and GAST, that are essential for organoid growth. By culturing human gastric stem cells in a medium containing EGF, NOG, RSPO, WNT, FGF10, GAST, and a TGF-β inhibitor, they facilitated the growth of gastric organoids with cystic bodies, derived from gastric glands or individual cells. Moreover, the organoids expressed the stem cell marker LGR 5 and gastric epithelial markers. 144
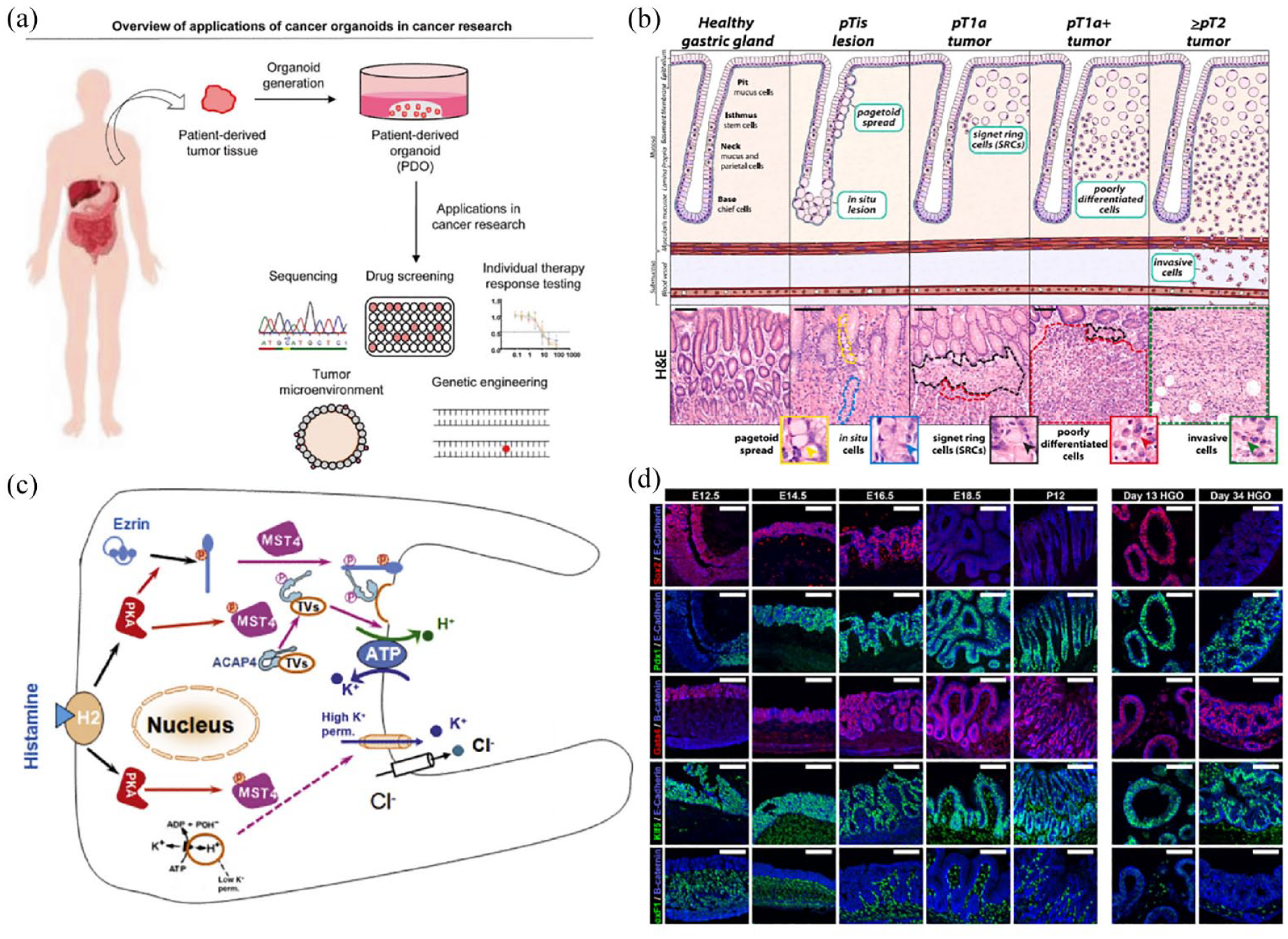
Application of gastric cancer organoids. (a) Preparation process of the tumor organoids. Adapted from Seidlitz and Stange. 142 (b) Histological characterization of diffuse-type gastric cancer (DGC) progression. Adapted from Monster et al. 145 (c) Mechanisms of parietal cell acid secretion. Adapted from Yao and Smolka. 146 (d) Human gastric organoids recapitulate normal antrum development of mouse embryos. Adapted from McCracken et al. 147
One factor that impacts gastric carcinogenesis is Helicobacter pylori infection. 148 Accordingly, an organoid model of H. pylori infection has been developed by microinjecting H. pylori into a human gastric organoid model to study the pathogenesis of GC. This model leads to enhanced gastric epithelial cell proliferation within 24 h and recapitulates normal embryonic development; the molecular and morphogenetic processes that occur during antrum development are conserved between rodents and humans (Figure 7D). 147 Furthermore, the induction of epithelial proliferation was investigated by infecting human and murine gastric organoids with H. pylori and examining the role of CD44. In response to H. pylori infection, lost epithelial proliferation from infected organoids derived from the stomach of CD44-deficient mice. Human gastric fundus organoids exhibited induced proliferation upon infection with H. pylori, which was not seen in organoids pretreated with CD44-specific peptide inhibitors, suggesting that CD44 plays an important role in H. pylori-induced epithelial cell proliferation. 149 However, the precise behavior of H. pylori within host cells remains unclear. Most H. pylori are typically observed near the pit area in human biopsy samples. Thus, using human adult stem cell-derived gastric organoids and single-cell RNA-seq, Aguilar et al. 150 found that H. pylori preferentially binds to large and granular gastric pit cells. This preference is attributed to bacterial chemotaxis to host urea. H. pylori also induces PD-L1 expression on gastric epithelial cells, promoting cell proliferation and survival. The interaction between PD-L1 and PD1 on the surface of cytotoxic T lymphocytes (CTLs) fails to induce apoptosis. To elucidate this specific mechanism, Holokai et al. 151 co-cultured H. pylori-infected human gastric organoids with host immune cells and treated them with a PD-1 inhibitor. The expression of PD-L1 by H. pylori inhibited CTL proliferation, however, in the co-cultured organoids, CTL proliferation increased, and the expression of PD-1 on gastric epithelial cells decreased. Studies have suggested that H pylori’s ability to reduce proton pump expression allows it to regulate gastric luminal acidity and also affects parietal cell organelle interactions involved in histamine-induced acid secretion (Figure 7(c)). 146 Hence, H. pylori-infected organoids provide a valuable tool for studying the interaction between gastric epithelial cells and H. pylori, shedding light on the relationship between H. pylori and GC.
The Cancer Genome Atlas (TCGA) categorizes GC into four molecular subtypes: Epstein-Barr virus-positive, microsatellite instability, chromosomal instability, and genomic stability. 152 To assess the response of conventional drugs in GC organoids, Seidlitz et al. 153 established 20 human GC organoids, including DD107 (body cancer), DD109 (AEGI), DD191 (AEG II), and DD282 (antral cancer). Their analysis revealed variations in drug resistance, with DD109 and DD191 showing resistance to 5-FU and epirubicin. DD109 responded well to irinotecan treatment, whereas DD107 only elicited a response at higher concentrations. Moreover, whole-genome sequencing matched DD191 and DD282 to the MSI subtype, DD107 to the GS subtype, and DD109 to the CIN subtype. Collectively, this data aid in selecting suitable drugs for patients through tumor organoid screening.
To investigate the similarities and differences between GC organoids and primary tumors, Kumar et al. 154 compared scRNA-seq data sets from GC organoids and primary tumors. The analysis revealed sublineage heterogeneity within GC organoids, while identifying module and genes associated with GC compared to normal gastric organoids. Additionally, tumor-associated epithelial cells exhibited increased transcriptional plasticity, suggesting that GC organoids can be used as an in vitro model to study GC transcriptional plasticity. Interestingly, H. pylori-infected GC exhibited independent liver cancer growth factor and tumor necrosis factor, which can affect the development and invasiveness of GC. 155 Organoids also provide insights regarding the pathogenesis of diffuse GC (Figure 7(b)). 145 Therefore, GC organoids hold great potential as a model for studying GC mechanisms and related drugs.
CRC organoids
CRC accounts for 6.0% of all annual cancer diagnoses worldwide, with a mortality rate of 5.8%. 2 Approximately 20% of newly diagnosed CRC patients will develop synchronous liver metastases (LM), and at least half of the patients with postoperative metastatic disease will develop liver metastases. 156 Mo et al. 157 constructed a live biobank of 50 CDC liver metastasis (CRLM) organoids derived from primary tumors and paired liver metastatic lesions (Figure 8(a)), confirming that the histopathological structure of CRLM organoids retain that of the parental tumors and recapitulate the genomic map of the corresponding patient’s tumors. Betge et al. 41 further generated patient-derived organoids to establish CRC organoids from 13 patients to understand their morphological diversity via image analysis (Figure 8(d)). Meanwhile, Wang et al. 67 utilized single-cell RNA-seq analysis of organoids derived from seven patients with CRC and found that CRC organoids exhibited a higher proportion of stem-like cells and retained the genome of in vivo tissues, global DNA methylation levels, CNV, and point mutations observed in patient tumor samples. Additionally, they identified genes closely related to the progression and metastasis of CRC, including PROCR, SCD, BMP 4, CEACAM6, TESC, and TGFBI.

Application of colorectal cancer organoids. (a) A living biobank of CRLM organoids. Adapted from Mo et al. 157 (b) Colon organoids stably expressing GFP were cultured alone or together with EPCAM-CAR cells or parental NK-92 cells that were labeled by anti-CD45 staining. Adapted from Schnalzger et al. 158 (c) Induction of tumor reactivity in circulating T cells by co-culture with autologous tumor organoids. Adapted from Dijkstraet al. 64 (d) Image-based profiling captures the phenotype diversity of patient-derived cancer organoids. Adapted from Betge et al. 41
The combination of organoids and CRISPR/Cas9 technology allows one to study the molecular mechanisms underlying CRC. For instance, Matano et al. 159 successfully established organoids with knockout mutations in key genes (APC, TP53, SMAD4, KRAS, and PIK3CA) using CRISPR/Cas9 technology and validated their effects through niche factor experiments. This approach not only validates known genes but also facilitates the identification and functional characterization of novel driver genes in CRC. For example, Li et al. 160 used genome-wide CRISPR screening to identify LGALS2 as a gene involved in regulating cellular response to oxidative stress in CRC. Meanwhile, Ringel et al. 161 combined CRISPR with organoids to confirm genes implicated in TGF-β mediated resistance to growth restriction. Therefore, the integration of organoids and CRISPR technology enhances our understanding of the functional aspects of CRC pathogenesis.
Immunotherapy offers a promising approach to overcome cancer resistance by enhancing the patient’s immune system. It has also shown potential in the treatment of CRC. Personalized immunotherapy treatment relies on studying cell-cell communication and signaling within the TME. CAFs play an important role in CRC progression. Studies have identified pericryptal Lepr-lineage cells that proliferate and give rise to MCAM CAF, contributing to the formation of an immune-friendly TME. 162 However, the lack of CAFs in tumor organoids hinders effective drug screening and personalized treatment. Luo et al. 163 addressed this issue by co-culturing CRC PDOs with patient-derived CAFs in a 3D hyaluronan-gelatin hydrogel instead of conventional Matrigel. They performed RNA and whole-exome sequencing on the model and found that it retained the molecular characteristics of the patient’s tumor. The model also facilitated the evaluation of standard-of-care drugs. In addition to CAF co-culture, peripheral blood lymphocytes can be co-cultured with organoids. This approach can enrich tumor-reactive T cells derived from the peripheral blood of patients with mismatch repair-deficient CRC (Figure 8(c)). 64 Moreover, it allows for the prediction of immunotherapy sensitivity at different stages of treatment. 64 Chimeric antigen receptor (CAR)-engineered lymphocytes have demonstrated encouraging results; in leukemia CAR-NK-92 cell cytotoxicity was better monitored by growing colonic organoids together with epithelial antigen (EPCAM)-CAR cells or parental NK-45 cells (Figure 8(b)). 158 Therefore, this co-culture model provides valuable insights into immunotherapy mechanisms and enhances our understanding of how to improve treatment efficiency.
EC organoids
EC is one of the most dangerous malignancies in humans, with a global incidence rate of 3.1% and a mortality rate of 5.5%. EC is histologically classified into two types: esophageal squamous cell carcinoma (ESCC) and esophageal adenocarcinoma (EAC). 2 During embryogenesis, the esophagus is initially lined by ciliary columnar cells, which are replaced by stratified SCs as the maturation process progresses (Figure 9(b)). 164 Due to its specific anatomical location and structure, EC is often diagnosed at an advanced stage, limiting treatment options. Therefore, it is crucial to improve our understanding of the underlying mechanisms of both types of EC to achieve earlier and more accurate diagnosis. Fortunately, tumor organoid models have been established to study ESCC and EAC, providing valuable tools for further research in EC.46,165 Ko et al. 166 designed culture methods for mouse esophageal organoids and transgenic esophageal organoids using viral transduction (Figure 9(a and c)). For EAC, organoids derived from endoscopic biopsies of patients accurately reflect the histological characteristics of the tumor. Thus, subsequent whole-exome sequencing of these organoids revealed the expression of tumor-associated genomic features. Interestingly, different sources of EC organoids exhibit different responses to the same chemotherapeutic agent, facilitating personalized treatment plans.142,153,167 Freshly excised EAC tissue samples can also be used to culture organoid models, which represent the driver genes associated with primary tumors. These organoids exhibit similar sensitivities to various chemotherapy agents, including EGFR and MEK inhibitors. 46 This improved understanding of EAC holds promise for improving personalized treatment strategies.
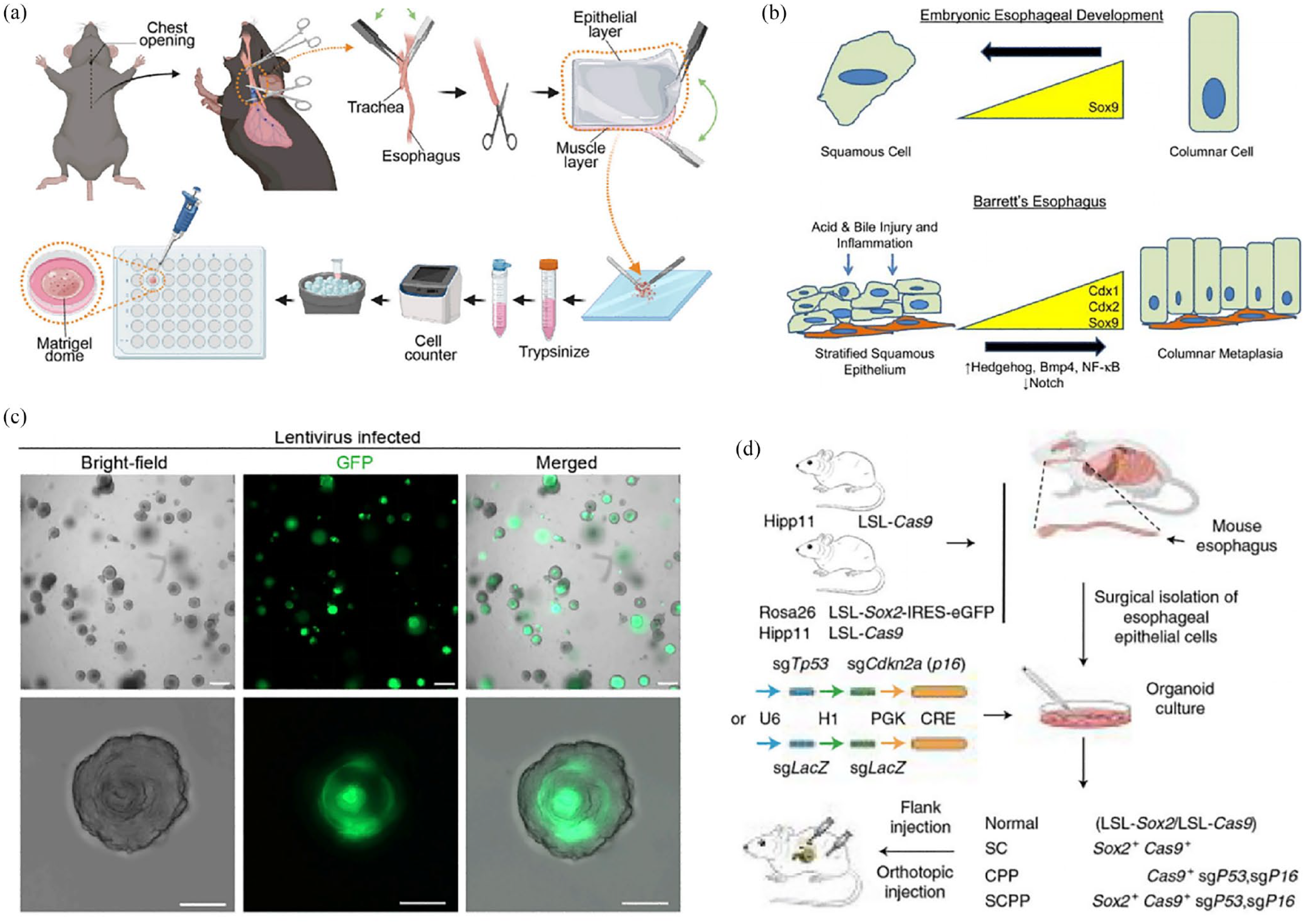
Application of EC organoids. (a) Schematic process of murine esophageal organoid culture. Adapted from Ko et al. 166 (b) Phenotypic changes in esophageal epithelium occur during normal development and Barrett’s esophagus. Adapted from Wang and Souza. 164 (c) Lentivirus (pEGIP)-transduced murine esophageal organoids. Adapted from Ko et al. 166 (d) Schematic diagram of generating organoids with different genotypes from mouse esophageal epithelial cells that were isolated from Rosa26CAG-loxp-stop-loxp-Sox2-IRES-Egfp (LSL-Sox2); H11CAG-loxp-stop-loxp-Cas9 (LSL-Cos9) mice. Adapted from Wu et al. 168
Regarding squamous cell carcinoma organoids, Kijima et al. 45 conducted paired organoid biopsies from patient tumors and adjacent normal mucosa. The associated organoids were cultured in Matrigel supplemented with DMEM/F12. The results demonstrated that organoids derived from tumor tissue exhibited atypical cells with high nuclear/cytoplasmic ratios and accumulated mutant P53 protein, while those derived from normal mucosa maintained normal nuclear features. Additionally, ESCC organoids express CD44 on the cell surface, similar to xenograft tumors. High levels of CD44 expression are associated with resistance to 5-fluorouracil and increased autophagy, providing enhanced protection against treatment. 45 To further understand the potential pathogenic alterations of the microbiome in esophageal neoplasia and which microorganisms represent risk factors for ESCC progression, Flashner et al. 169 co-cultured patient-derived esophageal organoids with the microbiome, which addresses the role of the bacterial microbiome in oesophageal homeostasis. Meanwhile, Wu et al. 168 generated mice with Cre-inducible Sox2 and Cas9 expression (Figure 9(d)), to generate an organoid that separates the normal and studied esophageal epithelium and transforms into ESCC by gene editing (including Sox2 overexpression), this validates that Sox 2 overexpression is able to promote ESCC transformation. As such, EC organoids offer a valuable platform for studying cancer treatment approaches and investigating mechanisms of therapeutic resistance.
Pancreatic cancer organoids
The incidence and mortality of pancreatic cancer have steadily increased over the past few decades, primarily due to challenges in early diagnosis and limited treatment effectiveness. The most common type of pancreatic cancer is PDAC, 170 and the most common mutation associated with PDAC is the activating KRAS mutation. 171 The establishment of pancreatic ductal adenocarcinoma organoids is typically achieved by collecting tumor tissues from patients (Figure 10(b)), which are then used for drug screening to predict patient response to chemotherapy and elucidate the associated mechanisms (Figure 10(a and c)).172–174 For instance, Tsai et al. 63 cultured organoids from the tumor tissues of 28 patients with pancreatic cancer, and performed immunohistochemical and immunofluorescent evaluation to characterize organoid expression of tumor markers Maspin, Muc5ac, and CRR9, pancreatic marker PDX1, and gastroenteropancreatic and hepatobiliary epithelial marker CK19 (Figure 10(d)). They found that pancreatic cancer tumor organoids have similar histological features at primary tumors. To further investigate the mechanisms involved in pancreatic ductal carcinoma, Boj et al. 19 established PDAC organoids that exhibit highly aneuploid karyotypes and express PDA-related biomarkers, such as CA19-9. Importantly, KRAS mutations were also found in these models. Hence, this model is beneficial for simulating the development of pancreatic cancer and maintaining primary tumor histopathology. 175
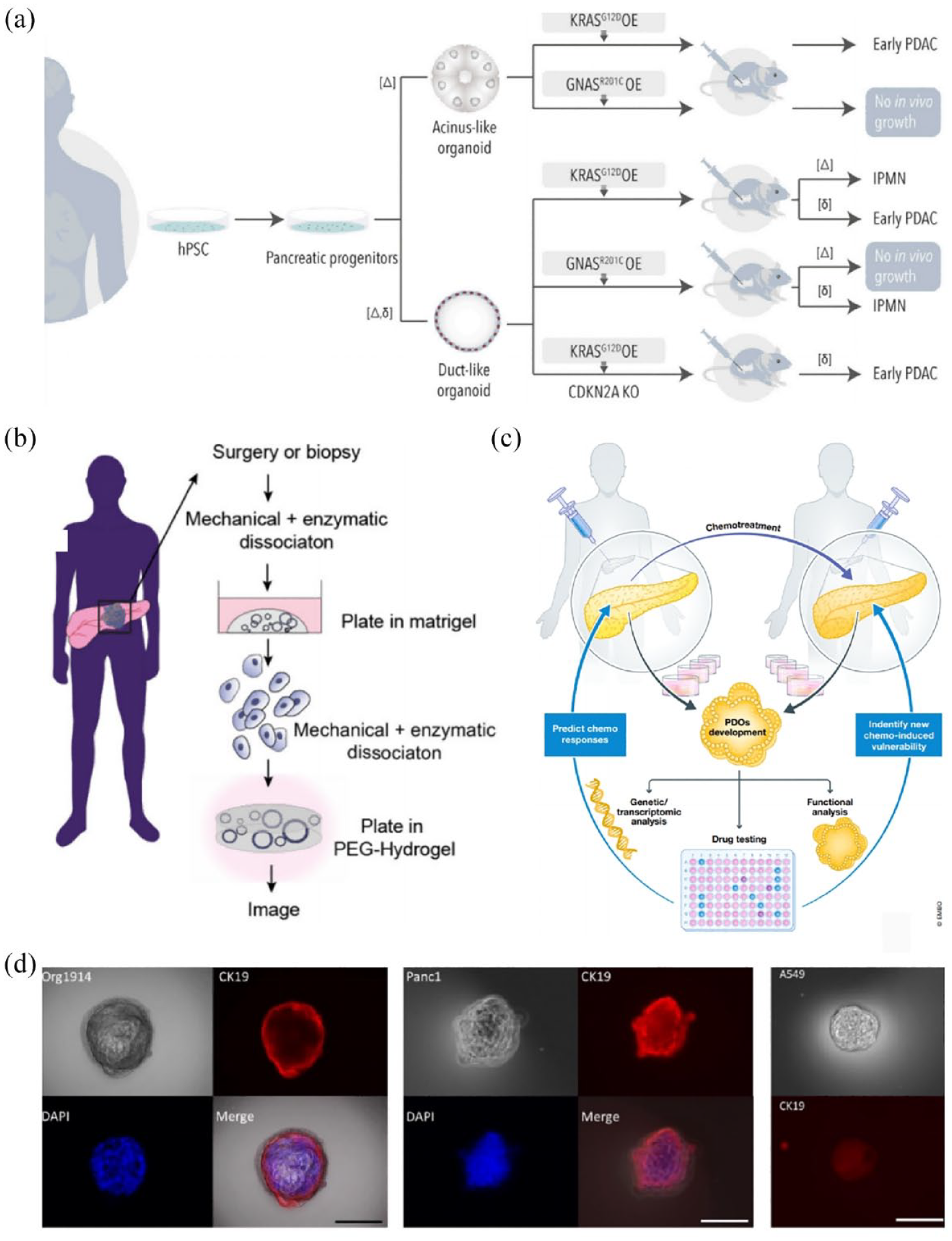
Application of Pancreatic cancer organoids. (a) Pancreatic cancer organoids used to study the role of PDAC driver mutations. Adapted from Casamitjana et al. 172 (b) Schematic of human pancreatic ductal organoid (hPDO) establishment. Adapted from Below et al. 174 (c) PDOs, a model to predict the patient response to chemotherapy and define new treatment-induced vulnerability. Adapted from Sandhya et al. 173 (d) Immunofluorescent Characterization of Organoids. Adapted from Tsai et al. 63
Additionally, PDAC organoids demonstrate similar drug sensitivity to patients with PDAC. Indeed, a retrospective clinical follow-up of patients treated with five conventional chemotherapy drugs (gemcitabine, nab-paclitaxel, irinotecan (SN-38), 5-fluorouracil (5-FU), and oxaliplatin) revealed that PDAC organoids exhibit responses similar to those of the patients. Hence, the efficacy of chemotherapy drugs can be assessed by organoids. 176 By analyzing the area under the concentration-time curve (AUC) of individual drugs in PDAC organoids, with an AUC < 0.56 indicating sensitivity and an AUC ⩾ 0.56 indicating resistance, ineffective drugs can be eliminated, and the optimal treatment strategy can be identified. 175 Interestingly, organoids also facilitate the discovery of novel targets and chemotherapeutic agents to overcome the resistance of pancreatic cancer to conventional drugs. For example, the establishment of a PDAC organoid biobank led Zhou et al. 177 to discover that irbesartan improves the sensitivity of PDAC to chemotherapeutic drugs by inhibiting the Hippo/YAP1 pathway and reducing c-Jun expression. Therefore, combining chemotherapeutic drugs with irbesartan could improve therapeutic efficacy.
The investigation of drug resistance in pancreatic cancer involves not only relevant pathways but also the TME. Koikawa et al. 178 found that Pin1 is overexpressed in CAFs and cancer cells in human PDAC. Subsequently, through indirect co-culture of PDAC organoids with CAFs, they found that Pin1 promotes the hyperplastic and neoplastic TME, enhancing the growth of PDAC in organoids and related cancer phenotypes to better simulate the TME of PDAC. Additionally, Below et al. 174 established pancreatic organoids using PEG CBF-0.5 hydrogel, which recapitulated the growth and polarization observed in organoids cultured in Matrigel. Importantly, the stromal cell population in pancreatic tumors was retained, exhibiting similar phenotypes and signaling as observed in vivo. This model allows for a deeper understanding of the mechanisms underlying drug resistance in PDAC within the context of the TME. In conclusion, PDAC organoids hold great potential in cancer drug screening, elucidating drug resistance mechanisms, and the research and development of related cancer therapies.
Liver cancer organoids
Primary liver cancer (PLC) is the second most fatal malignancy globally. 2 The three most common subtypes of PLC include HCC, ICC, and hepatocellular-cholangiocarcinoma (HCC/CC), with HCC being the most prevalent. 179 Liver cancer organoids can be used to understand liver development and its function under healthy conditions, as well as the pathogenesis of related diseases, including the mechanism of HCC development induced by HBV (Figure 11(a and (b)).54,180 Tumor organoid models preserve the histological structure, gene expression, and genomic landscape of the original tumor. For example, Broutier et al. 44 established organoids derived from eight individuals with PLC, representing the three subtypes. Through marker analysis, histological evaluation, and whole-genome transcriptome (RNA-seq) analysis, they verified that these organoids retained the histological characteristics, marker expression, and transcriptomic alterations of the corresponding patient tumor subtypes.

Application of Liver cancer organoids. (a) Liver organoids are used in personalized medicine. Adapted from Prior et al. 180 (b) Patient-derived organoids from HBV+ patients. Adapted from Dong et al. 54 (c) The life cycle of the HBV. Adapted from Dong et al. 54 (d) HBV infection in vitro was infected with human liver organoids. Adapted from De Crignis et al. 181
One predisposing factor for liver cancer is HBV infection. 182 Liver organoids provide a valuable platform for studying the complete life cycle of HBV and investigating the distinct functions of viral proteins during the infection process (Figure 11(c)), 54 as well as the mechanisms underlying HBV infection (Figure 11(d)). De Crignis et al. 181 employed transcriptomic analysis of HBV-infected liver organoids derived from non-tumor cirrhotic tissues of patients following liver transplantation. They identified early gene markers (ADAMTS1, CCNA2, CORO5A, PTHLH, STMN4, and STY5) of liver cancer and validated its proximity to genes associated with HCC in TCGA Liver Hepatocellular Carcinoma database. Meanwhile, Chan et al. 183 established HCC organoids and discovered that silencing protein methyltransferase 6 (PRMT6) promotes tumorigenesis. Further investigations revealed that PRMT6 interacts with arginine 100 of CRAF, reducing its RAS binding potential and altering the MEK/ERK signaling pathway. Similarly, Li et al. 184 employed HCC organoids to determine that kinesin family member 15 (KIF15) promotes the cancer stem cell (CSC) phenotype and malignancy.
Drug resistance is a major factor contributing to the high mortality rates among patients with HCC, yet its mechanism remains poorly understood. Wang et al. 185 found that elevated expression of Frizzled-10 (FZD10) promotes hepatic CSC expansion and lunvalatinib resistance. Further validation using tumor organoids showed that FZD10 can be used to predict drug resistance. Lampis et al. 186 established cholangiocarcinoma PDOs and demonstrated that miRNA21 mediates cholangiocarcinoma (CCA) cell resistance to HSP90 inhibitors by downregulating DNAJB90. This suggests that HSP 90 inhibitors may be developed for treatomg CCA. Sinha et al. 187 confirmed that targeting the 12-HHTrE-LTB4R2-CTNNB1-YAP1 pathway initiated by hepatic stellate cells (aHSCs) in HCC organoids represented a potential therapeutic strategy for HCC. Moreover, Li et al. 188 established 27 liver cancer organoids and assessed their response to 129 anti-cancer drugs, revealing that most drugs were either ineffective or exhibited specific organoid activity, while others, including histone deacetylase inhibitors, proteasome inhibitors, DNA topoisomerase II inhibitors, protein translation inhibitors, and RNA synthesis inhibitors, demonstrated broad effectiveness.
Gastroenteropancreatic neuroendocrine neoplasm organoids
Gastroenteropancreatic neuroendocrine neoplasms (GEP-NENs) refer to a group of poorly understood diseases, categorized into two subtypes: gastroenteropancreatic neuroendocrine tumors (GEP-NET) and gastroenteropancreatic neuroendocrine carcinoma (GEP-NECs). 189 Unfortunately, surgical intervention remains the only treatment option for these diseases, and the prognosis is unfavorable with a rising incidence. 190 Moreover, GEP-NENs are heterogeneous malignancies; thus, a suitable model is urgently needed to understand their pathogenesis and conduct drug screening. D’Agosto et al. 191 successfully established long-term cultures of G2 metastatic small intestinal NET organoids, preserving the key genetic drivers and endocrine cell lineage expression. This model not only facilitates drug screening but also advances our understanding of the biology of G2 metastatic small intestinal NET.
In the case of GEP-NEC, limited treatment options and high relapse rates pose significant challenges, compounded by a scarcity of studies. To gain further insights into the mechanisms of GEP-NEC development and drug reactivity, Dijkstra et al. 192 established GEP-NEC organoids, albeit with low establishment rates. Nevertheless, these organoids exhibited neuroendocrine markers and demonstrated susceptibility to cisplatin and everolimus. Thus, the establishment of numerous organoid models holds promise for comprehensive analysis of the evolving biological behavior and drug sensitivity during GEP-NEC progression.
To further understand GEP-NENs, Kawasaki et al. 20 established 25 GEP-NENs organoids and conducted molecular and CRISPR gene-editing studies. Whole-genome and whole-exome sequencing confirmed the preservation of gene status and neuroendocrine phenotype in the organoids. The in vitro drug testing results were consistent with clinical recommendations, affirming the utility of organoids for drug screening. Furthermore, organoid whole-genome sequencing and RNA-seq data analysis revealed that the characteristic manifestation of GEP-NEN is due to numerous chromosomal aberrations and frequent RB1 alterations. Finally, by introducing a TR gene mutation and over-expressing NE-transcription factor (NE-TF), it was demonstrated that TP53 and RB1-double knockout (TR-KO) organoids generated through CRISPR-Cas9 can synergistically induce cell reprogramming into the NE lineage. Taken together, these findings emphasize the unique contribution of organoids as preclinical research models for mechanistic investigations of GEP-NENs.
Application of digestive tract tumor organoids
Understanding the development and progression of digestive tract tumors
The pathogenesis of many gastrointestinal tumors remains poorly understood, however, the advent of organoid models has facilitated their investigation. For example, in studying the pathogenesis of H. pylori-related gastric ulcers and GC, Idowu et al. 193 found that H. pylori inhibits IFN-γ signaling, induces inflammatory responses in neighboring epithelial cells, and causes DNA damage leading to carcinogenesis. Additionally, H. pylori infection increases the rate of DGC tumor cell proliferation and tumor progression. 145 Using CRC organoids, Li et al. 194 revealed that during CRC progression, SOX2 enhances aggressiveness, proliferation, and liver metastatic growth. Meanwhile, to investigate the mechanisms underlying pancreatic cancer cachexia, Vaes et al. 195 established pancreatic tumor organoids from eight patients with pancreatic cancer and observed variations in the expression of cachexia-related genes, including IL6, TNFA, IL8, IL1A, IL1A, MCP1, GDF15, and LIF. Interestingly, certain genes, such as IL1A and IL1B, exhibited decreased expression in organoids from cachexia and non-cachectic patients, while others like LIF, IL8, and GDF15 were upregulated. Notably, tumor organoids from patients with cachexia exhibited higher secretion levels of IL8 and GDF15. Establishing a human pancreatic tumor organoid biobank offers a platform for storing well-annotated cancer samples, facilitating a deeper understanding of the mechanisms that drive cancer cachexia.
Organoids can also be used to validate the influence of various factors on tumorigenesis. For example, Han et al. 196 investigated the effect of the aryl hydrocarbon receptor (AhR) on the response of colonic epithelial cells to IL-22. They found that IL-22 treatment induced the phosphorylation of STAT3, inhibiting organoid growth. Through intestinal cell-specific AhR-knockout organoids, they further verified that the loss of SOCS3 increased pSTAT3 levels and inhibited organoid growth, demonstrating that AhR signaling regulates the IL-22 response of colonic epithelial cells by regulating SOCS3 expression. In the same study examining the role of the mu-opioid receptor (MOR) in cancer progression and recurrence, increased MOR expression was observed in PC organoids and undifferentiated regions of resected PC tissue, suggesting its involvement in cancer progression. 197 Meanwhile, the pathogenesis of CCA associated with primary sclerosing cholangitis (PSC) remains largely unknown. Hence, CCA organoids were exposed to five cytokines (IL-1β, IL-6, IL-17A, interferon-γ, and TNF-α) and the effects on cell proliferation were assessed; only IL-17A had a proliferative effect on cells. However, further verification is needed to determine if IL-17A represents a potential target for treatment. 198 In summary, tumor organoids can provide valuable insights into disease pathogenesis and can contribute to the development of new drugs and prevention strategies.
Drug sensitivity screening and the development of novel drugs
Numerous studies have utilized large-scale tumor organoid models to conduct drug sensitivity screening and develop novel drugs for various gastrointestinal cancers. For example, Driehuis et al. 199 performed in vitro testing of 76 therapeutic agents using 30 PDOs from pancreatic and distal bile duct tumors. They found that the PRMT5 inhibitor EZP015556 could be used to target MTAP-negative tumors. Additionally, Bai et al. 200 screened anti-ICC drugs using mouse ICC organoids and cell lines, observing a promising inhibitory effect following treatment with combined Hinokitiol and Palbociclib, which requires further validation in clinical trials. Hirt et al. 201 established a human PDAC organoid biobank with 31 genetically distinct PDACs, and a screening of 1172 FDA-approved compounds identified 26 drugs that effectively inhibit PDAC organoids. Additionally, biobanks for GC, 153 liver cancer, 188 and CRC 202 have been successfully established, the utilization of which can assist patients in selecting the most suitable treatment.
Drug screening for digestive tract tumors can be enhanced by combining organoids with other techniques to improve screening efficiency. Du et al. 203 cultured organoids in 384-well plates and assessed cell viability after incubation with 2036 FDA-approved compounds. They successfully miniaturized the organoid system by optimizing the organoid culture conditions in 384 wells and simplifying the operating procedures in the 1536-well plate, thus improving the drug screening efficiency. CRISPR-Cas9 combined with HCC organoids demonstrated the effective inhibition of cancer cells by ifenprodil, which can be used as an adjuvant to sorafenib. Moreover, Deben et al. 204 employed machine vision and convolutional network machine learning methods, known as Organoid Brightfield Identification-based Therapy Screening, to enhance drug screening in tumor organoids. These studies highlight the potential of PDOs for large-scale drug screening, contributing to the advancement of precision medicine.
Tumor organoids can retain the heterogeneity and histological characteristics of tumors, similar to the parental genotype, and therefore can be used for the development of novel drugs for gastrointestinal cancer. In fact, Bian et al. 205 developed novel SOS1 degraders and verified their effect through CRC organoids, highlighting their potential as novel therapeutic agents for KRAS mutant CRC. Additionally, using CRC organoids, Zhao et al. 206 found that CSC and differentiated cancer cells (non-CSC) have unique metabolic characteristics, revealing that non-CSC-derived lactate enhances CSC organoid formation and tumor initiation. This finding provides new avenues for drug development.
Wang et al. 207 used CD44-positive HCC organoids to identify the efficacy of Hedgehog signaling inhibitors (GANT61) and found that they were more effective against HCC organoids compared to inhibitors targeting Notch, Hippo, or Wnt signaling. Moreover, combining GANT61 with sorafenib demonstrated a synergistic effect on CD44-positive HCC organoids, enhancing the effectiveness of sofranib treatment. Collectively, these studies show that the properties of the tumor organoids themselves facilitate the discovery of novel drugs.
Assessment of the treatment effect in patients
To minimize unnecessary side effects and delays in effective treatment caused by chemotherapy and radiotherapy, it is crucial to have appropriate preclinical models that can predict patient response to drugs. Tumor organoids, which retain the histological and molecular features of tumors, offer a promising solution for predicting the sensitivity of patients to chemoradiotherapy on an individual level. For example, Pasch et al. 208 established CRC organoids in five patients with CRC and monitored the organoid response to chemotherapy and radiation using optical metabolic imaging. They evaluated the drug’s efficacy in terms of diameter and optical redox ratio. Interestingly, they found that metastatic CRC organoids derived from neoadjuvant FOLFOX (5-FU, leucovorin calcium, and oxaliplatin) chemotherapy exhibited resistance to 5-FU but responded well to FOLFOX chemotherapy, consistent with clinical findings where the patient did not respond to 5-FU. Meanwhile, Yao et al. 209 generated a live organoid biobank to predict the chemoradiotherapy response of advanced rectal cancer in patients with locally advanced rectal cancer treated to neoadjuvant chemoradiotherapy (NACR). The results showed that the response of PDOs to chemical radiotherapy was closely matched to the actual response of the corresponding patients, with an accuracy of 84.43%, sensitivity of 78.01%, and specificity of 91.97%. Therefore, the PDO model can be used to predict the treatment effect of the patients. A similar study using patient-derived PDAC organoids was conducted by Frappart et al., 210 which validated the model for predicting drug response.
PDOs have also demonstrated their efficacy in predicting response outcomes after targeted therapy in patients with GC. 211 Comparing patient-derived PDOs with standard chemotherapy treatments, such as epirubicin, oxaliplatin, and 5-FU, the investigators found that each PDO was associated with the response shown by the patient after the treatment. GC organoids that demonstrate complete responses to chemotherapy are found to be sensitive to treatment, while those that do not respond to chemotherapy exhibit resistance. Moreover, following the transplantation of PDOs into immunodeficient mice, the resulting organoids were highly similar to the patient’s tumor in terms of histology, reinforcing the efficacy of PDOs in predicting treatment response.
In conclusion, PDOs have shown promise in predicting the drug response of patients to chemoradiotherapy, enabling clinicians to make timely adjustments to drug combinations, thus, reducing patient suffering. Therefore, organoids have the potential to be used as a companion diagnostic tool for gastrointestinal cancer treatment.
Immunotherapy
Recently, cancer immunotherapy has made significant breakthroughs in the field of immunology research due to its low toxicity and long-lasting efficacy. 212 Tumor organoids have also made progress in immunotherapy (Figure 12(a)). 213 Although whole ALI cancer organoid cultures and microfluidic cultures can be used to reconstitute the TME by preserving endogenous matrix components, co-culturing tumor organoids with relevant immune cells provides a better understanding of the interaction between epithelial and immune cells. For example, Wang et al. 214 co-cultured HCC organoids with either endothelial cells or fibroblasts, leading to the production of new blood vessel-related markers (VEGFR3, VE GF, and HIF-α), tumor-related inflammatory factors (CXCR2, CXCL4, and TNF-α), and molecules associated with induced epithelial-mesenchymal transition (TGF-β, Vimentin, and MMP-12). This co-culture favored the growth of HCC cells in organoids. Meanwhile, co-culturing PDAC organoids with CAFs was shown to promote the growth of tumor organoids. 215 Co-culture of mouse PDAC organoids with pancreatic stellate cells revealed the ability of CAFs to produce a desmoplastic matrix and identified new subsets of CAFs that secrete IL-6 and other inflammatory mediators. These findings provide strong evidence for the heterogeneity of CAFs in PDA tumor biology. 216

Tumor organoids used for immunotherapy and for mimicking the tumor microenvironment. (a) Tumor organoids used as personalized immunotherapy. Adapted from Yuki et al. 213 (b) Tumor organoids were co-cultured with T cells. Adapted from Cattaneo et al. 217 (c) Tumor organoids were co-cultured with immune cells to construct the immune microenvironment. Adapted from Xu et al. 218 (d) Retained fibroblasts in human and mouse tumor organoids. Adapted from Neal et al. 62
Cattaneo et al. 217 established a co-culture system with peripheral blood T cells that enriched reactive T cells and facilitated the assessment of the killing efficiency of T cells against tumor organoids (Figure 12(b)). In contrast, Tsai et al. 63 co-cultured CD3 T cells isolated from peripheral blood mononuclear cells (PBMCs) with pancreatic cancer organoids and found viable T cells expressing Ki67. Co-culturing of cholangiocarcinoma organoids with PBMCs or purified T cells revealed the toxic effects of T cells on most organoids. 219 Fibroblasts are also found in mouse and human tumor organoids (Figure 12(d)). 62 Another study showed that co-culturing PDA organoids with immune cells inhibited the effector function of T cells in the TME. 220 Polymorphonuclear-MDSCs inhibit CTL anti-tumor activity by depleting L-arginine and L-cysteine and generating ROS, rendering immunotherapy less effective. However, detargeting TBK1 can overcome immunotherapy resistance by enhancing the response to PD-1 blockade, lowering the cytotoxicity threshold of effector cytokines (TNF and IFN-γ). 221
Co-culturing tumor organoids with immune cells can simulate the TME (Figure 12(c)); the establishment of the PDO system using ALI, and the mock immune checkpoint blockade with anti-PD-1/anti-PD-L1 have been extensively reviewed elsewhere.62,218 Moreover, Teijeira et al. 222 have tested the therapeutic efficacy of CEA-CD3 T-cell engagers on colon cancer by co-cultivating tumor organoids, autologous fibroblasts, and T cells. However, additional PDO methods have been established, including primary tumor-derived organotypic cell clusters for evaluating cancer immunotherapy, 223 a microfluidic perfusion system (EVIDENT) for studying the interaction between tumor fragments and TILs, 224 and a microfluidic chip device developed by Haque et al. 225 to simulate the PDAC TME and observe the active crosstalk between cancer and stromal cells. This microfluidic enables accurate screening of immunotherapeutic drugs by better simulating the TME and observing the effects of stromal cells on chemotherapy efficacy. Moreover, it enables drugs targeting PSC or macrophages to significantly enhance the efficacy of chemotherapy drugs in killing tumor cells under TME modulators. Such effects cannot be replicated in tumor cell culture systems lacking stromal cells. Hence, this device has a superior ability to simulate the TME for accurate screening of immunotherapeutic drugs. Therefore, PDOs serve as a potent tool for adjuvant research in immunotherapy.
Conclusions and outlook
Compared with PDX, tumor organoids possess several advantages, such as maintaining high tumor cell heterogeneity, genetic fidelity, reduced time and cost, and the ability to better simulate the TME. This makes them a promising tool for various clinical applications as well as for the analysis of tumorigenesis and cancer progression, predicting drug responses, conducting high-throughput drug screening, and discovering novel anti-cancer drugs. Additionally, combining organ chip and CRISPR-Cas9 technologies with tumor organoids can further enhance their role.
Despite the potential of organoids in cancer research and clinical applications, certain limitations remain. For instance, the current organoid cultures lack important components, such as gut microbiota, vascular endothelial cells, neurons, and immune cells, which limits their ability to fully capture tumor physiology and pathology. 37 However, by incorporating co-cultures with immune cells and employing other methods, it is possible to better simulate the TME and improve the accuracy of immunotherapy.63,110,111 Although some progress has been made in these aspects, most organoids lack stromal cells, which can affect signal transmission and the ability to fully replicate patient responses to immunotherapy. This hinders the accurate clinical evaluation of the pharmacological effects of immunomodulatory agents. Furthermore, the use of animal-derived ECM like Matrigel in organoid cultures can impact drug screening and genetic screening outcomes. However, efforts are underway to develop alternative materials that reduce the reliance on animal substrates.89,97 Standardization of organoid culture methods and establishing a material library for selecting appropriate culture materials, along with robust quality control, are essential steps in advancing organoid research.
One particular challenge in simulating the structural features closely related to the function and development of real organs, especially in digestive tract tumor organoids, is the absence of vasculature. Researchers are actively working to address this issue and have made some progress. For example, a vascular organoid model was established by Low et al. 226 However, only a few types of organoids have been successfully vascularized, indicating that further research is needed. Moreover, the structure, cellular composition, and gene expression of established organoids exhibit variability, which poses obstacles to high-throughput drug screening.
As technology continues to advance, there is a growing expectation for improved digestive tract tumor organoids. These advancements will not only lead to reduced costs associated with organoid production but also address challenges such as simulating the TME and vascular system. Efforts are underway to develop alternative materials to replace Matrigel, optimize the culture system of tumor organoids, and minimize variations among cultured organoids. Additionally, by integrating organoids with new technologies, the efficiency of drug screening can be enhanced.
Footnotes
Data availability
No data was used for the research described in the article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was sponsored by the Science and Technology Program of Health Commission of Jiangxi Province (No. SKJP220210528) and the Education Science and Technology Research Project of Jiangxi Province of China (No. GJJ2201921).