
Abstract
Type 1 diabetes (T1D) results from the autoimmune destruction of pancreatic β cells, leading to lifelong insulin dependence and significant health complications. Human pluripotent stem cell-derived β cells (hPSC-β cells) have emerged as a promising therapeutic alternative for restoring endogenous insulin production; however, limitations such as functional immaturity, immune rejection, and biosafety concerns such as tumorigenic risk continue to hinder clinical application. Recent advances in gene editing technologies, particularly clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9), offer precise tools to enhance or correct hPSC-β cell performance by improving glucose-stimulated insulin secretion (GSIS), reducing immune rejection, and reducing biosafety concerns. This review explores gene editing strategies developed to overcome the key barriers in hPSC-β cell-based therapy for T1D. We highlight how genetic modifications enhance or correct β cell function, promote immune evasion, and reduce biosafety concerns through precise and clinically relevant engineering. Finally, we discuss the current landscape of clinical trials and future directions for translating gene-edited hPSC-β cells into curative treatments for T1D.
Keywords
Introduction
Type 1 diabetes (T1D) is an autoimmune disease characterized by the destruction of pancreatic β cells, which are responsible for insulin production. This leads to absolute insulin deficiency, resulting in loss of glycemic regulation.1–4 Without adequate insulin secretion, patients experience chronic hyperglycemia, which can cause severe complications such as cardiovascular disease, neuropathy, and nephropathy.5–7 Currently, T1D affects approximately 8.75 million people worldwide, with incidence rates increasing annually, particularly in children and young adults. Studies indicate that the prevalence of T1D is rising by about 3% each year, highlighting the urgent need for improved treatment strategies. 8
While exogenous insulin injection helps regulate blood glucose levels, it presents significant challenges. Patients often struggle to maintain stable glucose levels, which increases the risk of hypoglycemia and long-term complications associated with hyperglycemia. Additionally, insulin therapy does not address the underlying loss of β cells, meaning patients must rely on lifelong injections. 9 Although advancements have been made in blood glucose monitoring and insulin injection, only 21% of adults successfully meet the glycemic targets set by the American Diabetes Association (ADA). 10
In pursuit of a functional cure, islet transplantation has emerged as a promising approach. The Edmonton protocol, developed in the early 2000s, marked a significant advancement by enabling the transplantation of allogeneic cadaveric islets into the portal vein of recipients. 11 This method has demonstrated temporary insulin independence and improved glycemic control. However, several challenges hinder its widespread application. The primary limitation is the scarcity of suitable donor cells, making this treatment inaccessible to a large portion of T1D patients. Additionally, patients receiving islet transplants must take lifelong immunosuppressive drugs to prevent immune rejection, which introduces risks such as increased susceptibility to infections and adverse side effects. 12 One of the most recently clinically available allogeneic islet cell therapies, Lantidra (donislecel), was approved by the U.S. Food and Drug Administration (FDA) in 2023, and it also requires concomitant immunosuppressive regimens to maintain graft survival and function. The initial and subsequent infusions require a minimum of 5000 and 4500 islet equivalents (IEQ/kg), respectively. Despite its clinical promise, the therapy remains constrained by donor availability, representing a persistent limitation. 13
Since the early 2000s, efforts to overcome donor shortages have included the generation of insulin-producing β cells from pluripotent stem cells (PSCs). These human pluripotent stem cell-derived β cells (hPSC-β cells) represent a renewable and scalable source for cell replacement therapies. 14 However, using hPSC-β cells as viable therapeutics presents considerable challenges. Current hPSC-β cells often exhibit functional immaturity, characterized by suboptimal glucose-stimulated insulin secretion (GSIS) compared to native β cells.15,16 Post-transplantation, these cells are susceptible to functional immaturity, compromised viability, and immune rejection. 17 Additionally, concerns over tumorigenicity and immune safety further complicate clinical applications.18,19
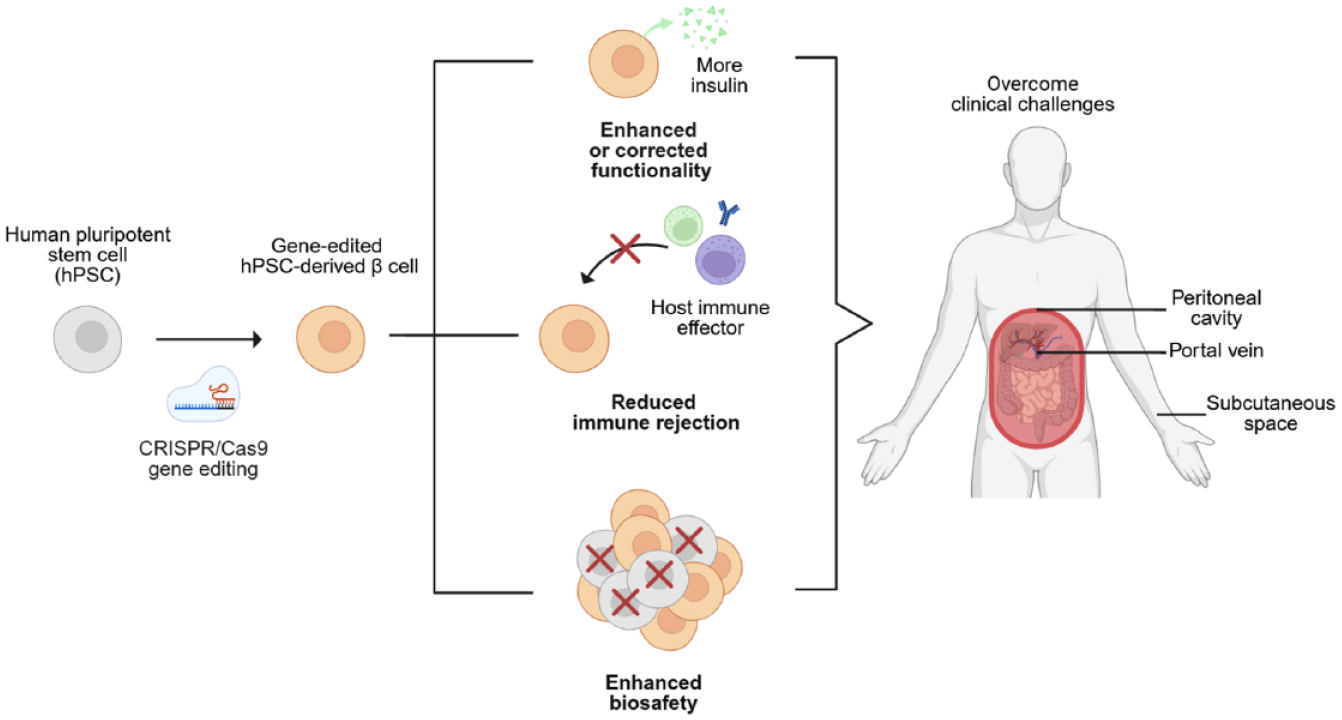
Recent advancements in gene editing tools such as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9) are enhancing the development of hPSC-β cells with improved functionality, immune evasion, and biosafety. 20 Precise gene modifications can enhance insulin secretion, reduce immunogenicity, and lower tumorigenic risk (Figure 1(a)). Here, we review recent progress and remaining challenges in developing gene-edited hPSC-β cells for the treatment of T1D. We focus on how advances in gene editing technologies, particularly CRISPR/Cas9, have been applied to overcome key barriers such as functional immaturity, immune rejection, and biosafety concerns. This review begins with an overview of hPSC-β cell differentiation protocols, followed by a discussion of strategies to enhance or correct GSIS, reduce immune responses, and improve biosafety (Table 1). The review also highlights recent clinical efforts to translate gene-edited hPSC-β cells into therapeutic products, with emphasis on multiplexed gene editing and tissue engineering approaches such as encapsulation system. By integrating insights from both basic and translational research, this review provides a comprehensive perspective on the therapeutic potential and future directions of gene-edited hPSC-β cells in regenerative medicine for T1D.
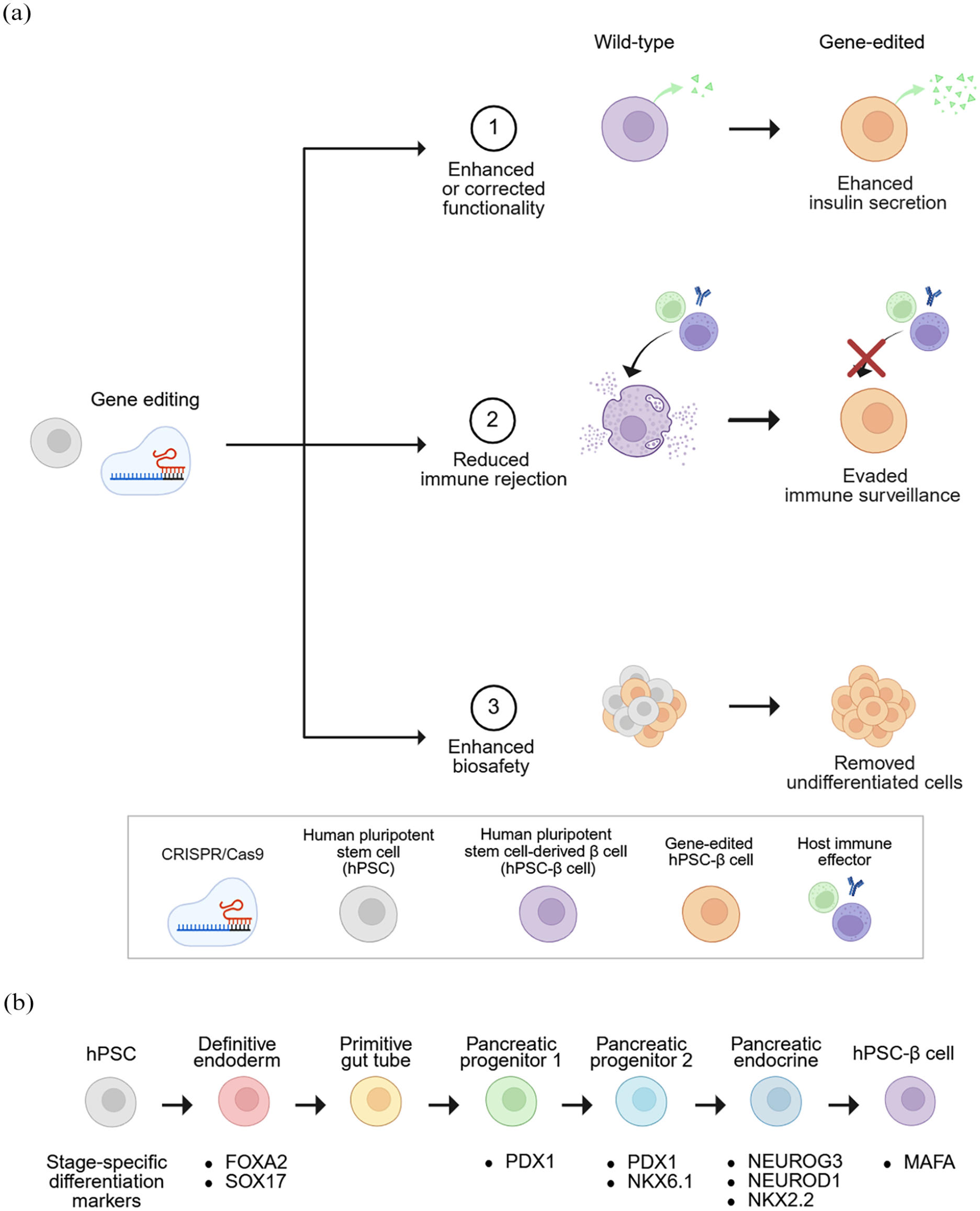
Overview of differentiation markers and gene editing strategies for generating functional hPSC-β cells: (a) CRISPR/Cas9-mediated gene editing of hPSCs followed by directed differentiation into hPSC-β cells, with modifications aimed at enhancing functionality and reducing immune rejection and biosafety concerns and (b) lineage-specific marker expression during stepwise differentiation to hPSC-β cells. All figures were created with BioRender.com.
Selected gene editing strategies for hPSC-β cells to enhance or correct functionality, reduce immune rejection, and enhance biosafety.
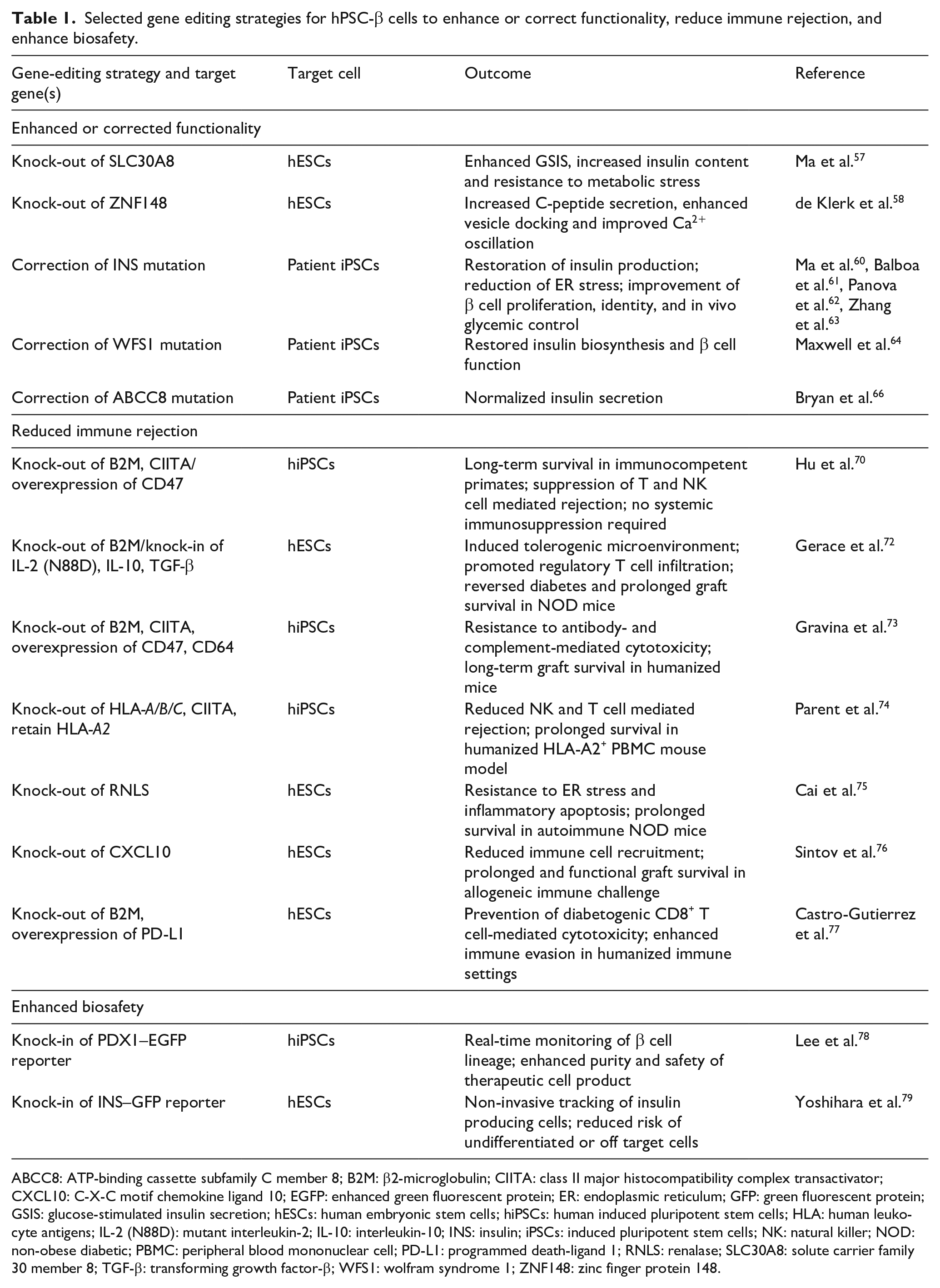
ABCC8: ATP-binding cassette subfamily C member 8; B2M: β2-microglobulin; CIITA: class II major histocompatibility complex transactivator; CXCL10: C-X-C motif chemokine ligand 10; EGFP: enhanced green fluorescent protein; ER: endoplasmic reticulum; GFP: green fluorescent protein; GSIS: glucose-stimulated insulin secretion; hESCs: human embryonic stem cells; hiPSCs: human induced pluripotent stem cells; HLA: human leukocyte antigens; IL-2 (N88D): mutant interleukin-2; IL-10: interleukin-10; INS: insulin; iPSCs: induced pluripotent stem cells; NK: natural killer; NOD: non-obese diabetic; PBMC: peripheral blood mononuclear cell; PD-L1: programmed death-ligand 1; RNLS: renalase; SLC30A8: solute carrier family 30 member 8; TGF-β: transforming growth factor-β; WFS1: wolfram syndrome 1; ZNF148: zinc finger protein 148.
Establishment and optimization of hPSC-β cell differentiation through culture system engineering
The development of human pluripotent stem cell-derived β cells (hPSC-β cells) was driven by the recognized need for a renewable source of insulin-producing cells to overcome the limitations of cadaveric islet transplantation. Early differentiation protocols successfully guided human pluripotent stem cells (hPSCs) through sequential stages mimicking embryonic pancreas development.21,22 The hPSCs, including embryonic stem cells (ESCs) and induced pluripotent stem cells (iPSCs), can be directed through a stepwise differentiation process into hPSC-β cells. This generally involves sequential stages of definitive endoderm induction, primitive gut tube formation, pancreatic progenitor specification, endocrine commitment, and finally β cell maturation (Figure 1(b)). These protocols directed hPSCs through key transcriptional milestones, beginning with the induction of definitive endoderm [marked by forkhead box A2 (FOXA2), sex-determining region Y-box 17 (SOX17)], followed by patterning into the primitive gut tube, and specification into pancreatic progenitors expressing pancreatic and duodenal homeobox 1 (PDX1). Subsequent exposure to stage-specific signaling cues led to the activation of NK homeobox 6.1 (NKX6.1), a critical determinant of β cell lineage commitment, and later, transient expression of neurogenin-3 (NEUROG3), which directs endocrine specification. This cascade further progresses with expression of neurogenic differentiation 1 (NEUROD1) and NK homeobox 2.2 (NKX2.2), culminating in the activation of v-Maf musculoaponeurotic fibrosarcoma oncogene homolog A (MAFA), a marker of β cell maturation. As these transcription factors are activated in a tightly regulated sequence, their temporal coordination is critical for proper β cell fate. Notably, premature expression of NEUROG3, a key regulator of endocrine specification, can lead to polyhormonal differentiation rather than the formation of monohormonal insulin-producing cells.16,23
Each stage of differentiation requires precise control of both signaling cues and culture conditions to ensure proper lineage progression. In particular, the transition from pancreatic progenitors to mature β cells remains challenging, as monolayer two-dimensional (2D) cultures often fail to support full maturation and robust glucose-responsive insulin secretion. In contrast, three-dimensional (3D) systems, including spinner flasks and transwell aggregates, have demonstrated enhanced endocrine induction and functional maturation, yet face significant challenges such as diffusion limitations including nutrient and oxygen gradients between the core and outer regions of spheroidal aggregates, scalability issues, and batch-to-batch variability.24,25 Nonetheless, a breakthrough was achieved with a 2D planar culture system, which demonstrated that modulating the mechanical properties of the cellular microenvironment, particularly stiffness, can enable β cell maturation in 2D conditions. 26 Specifically, monolayer culture on rigid tissue culture polystyrene was found to increase actin polymerization, which in turn repressed NEUROG3 expression and inhibited endocrine induction. This inhibition was overcome by transient cytoskeletal depolymerization using latrunculin A during the initial phase of endocrine induction, which restored NEUROG3 expression and enabled downstream activation of β cell genes. 27 The protocol was further optimized using 3,3′,5-triiodo-L-thyronine (T3), activin receptor-like kinase 5 inhibitor II (ALK5 inhibitor II), smoothened antagonist 1 (SANT1), and retinoic acid, followed by maturation in enriched serum-free medium. This planar methodology enhances scalability, accessibility, and standardization. However, the resulting hPSC-β cells still exhibit immature insulin secretion capacity compared to native human islets, highlighting the need for further functional optimization.28,29
Challenges in function of hPSC-β cell
Although hPSC-β cell technologies have advanced significantly in recent years, several critical barriers still hinder their clinical translation. Key challenges include functional immaturity, immunogenicity, and tumorigenicity associated with the use of hPSCs, each of which arises from distinct cellular and molecular mechanisms. Gene editing has emerged as a promising strategy to address these limitations. One of the most fundamental limitations is the inability of hPSC-β cells to recapitulate the dynamic insulin secretory capacity of native pancreatic β cells. Although they are capable of synthesizing and storing insulin, their glucose-stimulated insulin secretion (GSIS) remains suboptimal. Human primary islets typically exhibit a GSIS index of 3–5, whereas hPSC-β cells generally show a lower index of around 1–2. 26 While in vivo maturation of hPSC-derived pancreatic endoderm cells (PECs) has been clinically explored, their functional performance remains markedly inferior to that of native human islets. 30 In a phase 1/2 trial, subcutaneously implanted PECs led to detectable meal-responsive C-peptide secretion by 26 weeks; however, peak stimulated levels were only 1%–2% of those required for insulin independence. Explanted grafts showed limited numbers of insulin-positive cells with partial expression of β cell maturity markers such as MAFA, and an unfavorable insulin-to-glucagon cell ratio. Moreover, C-peptide responses were delayed, peaking at 90–180 min post-meal. These findings indicate that in vivo maturation alone is insufficient to overcome the intrinsic functional immaturity of hPSC-β cells. This reflects the incomplete recapitulation of hPSC-β cell maturation, which remains limited by intrinsic transcriptional and functional immaturity. 31
Immature insulin secretory function in hPSC-β cells is frequently attributed to the dysregulated expression of transcription factors essential for endocrine differentiation, including NEUROG3, PDX1, and NKX6.1.32–34 In particular, aberrant NEUROG3 dynamics such as premature or excessive expression have been linked to the generation of polyhormonal cells co-expressing insulin, glucagon, and somatostatin, a feature characteristic of fetal or mis-specified islet cells. 35 The PDX1 transcription factor functions as a master regulator of pancreatic development and insulin gene transcription; however, in hPSC-β cells, its expression levels and nuclear localization are often inconsistent, which may underlie its limited ability to activate downstream β cell-specific target genes. 36 In particular, NKX6.1 plays a crucial role in β cell fate determination and proliferation, and its dysregulation along with other key transcription factors can lead to the generation of polyhormonal or non-β endocrine cells. 16 Additionally, altered expressions or activity of ion channels [e.g. KATP, adenosine triphosphate (ATP)-sensitive potassium channel] and zinc transporter 8 (ZnT8), which are essential for stimulus-secretion coupling, further changes the capacity of these cells to mount a rapid insulin response upon glucose elevation.37,38 The combined effects of incomplete transcriptional programing, suboptimal metabolic coupling, and impaired electrophysiological responsiveness significantly compromise the ability of hPSC-β cells to recapitulate the precise, glucose-dependent insulin secretory dynamics characteristic of mature human pancreatic β cells. Correction of these deficits necessitates the integration of refined differentiation protocols with targeted genomic interventions to reconstruct the regulatory networks essential for hPSC-β cell maturation and function.
Challenges in immune rejection of hPSC-β cell
Immune rejection poses a major barrier to the clinical application of hPSC-β cell transplantation, as these cells express polymorphic human leukocyte antigens (HLAs) that are readily recognized by the host’s immune system as non-self. 39 Upon transplantation, hPSC-β cells become targets of both direct and indirect T cell-mediated immune responses; briefly, host CD8⁺ cytotoxic T lymphocytes recognize allo-HLA class I molecules and induce cell death via perforin and granzyme release, while CD4⁺ helper T cells respond to HLA-derived peptides presented by antigen-presenting cells, amplifying inflammation through cytokine secretion and recruitment of additional immune effectors.40,41 Inflammatory cytokines such as interferon-γ (IFN-γ) and tumor necrosis factor-α (TNF-α) exacerbate immunogenicity by upregulating major histocompatibility complex (MHC) expression, adhesion molecules like intercellular adhesion molecule 1 (ICAM-1), and C-X-C motif chemokine ligand 10 (CXCL10) in the graft, further enhancing immune infiltration. While knock-out of HLA class I molecules can reduce T cell recognition, this approach often leads to increased vulnerability to natural killer (NK) cell-mediated cytotoxicity, as the absence of HLA triggers a “missing-self” response.42–44
To overcome immune rejection, clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 (Cas9)-based gene editing strategies have broadly focused on eliminating alloantigen presentation, modulating innate immune checkpoints, and suppressing pro-inflammatory signaling. These approaches aim to reduce both T cell- and NK cell-mediated cytotoxicity, often through combinatorial knock-out and knock-in designs. 41 In parallel, tissue engineering strategies such as encapsulation have been explored to achieve physical isolation of transplanted hPSC-β cells from the host immune system. While gene editing provides molecular stealth at the cellular level, encapsulation offers an immunoprotective barrier independent of cell modification, and both strategies are being developed to reduce reliance on systemic immunosuppression. However, despite its protective intent, foreign body responses are often initiated at the implant site, leading to macrophage activation, fibroblast recruitment, and ultimately fibrotic encapsulation of the graft. 45 This fibrotic barrier significantly impairs oxygen and nutrient exchange, causing hypoxia-induced stress and apoptosis within the hPSC-β cell cluster.30,46 Moreover, while encapsulation prevents immune cell infiltration, it does not block the diffusion of pro-inflammatory cytokines secreted by immune cells in the surrounding tissue, further contributing to hPSC-β cell dysfunction.
For patients receiving unencapsulated grafts, systemic immunosuppressive therapy is often necessary, yet this approach brings significant clinical risk, including heightened susceptibility to infections, increased cancer risk due to impaired immune surveillance, and metabolic side effects such as nephrotoxicity and insulin resistance. 47 Collectively, these multifaceted immunological pressures involving both innate and adaptive responses, as well as the physical and inflammatory consequences of biomaterial use, severely limit the persistence, function, and therapeutic efficacy of hPSC-β cell grafts in vivo.
Challenges in biosafety concerns of hPSC-β cell
The clinical application of hPSC-β cells requires careful consideration of biosafety aspects, particularly the potential risk of teratoma formation arising from residual undifferentiated hPSCs. 48 These cells express core pluripotency factors such as octamer-binding transcription factor 4 (OCT4), sex-determining region Y-box 2 (SOX2), and homeobox protein NANOG (NANOG), and may retain proliferative potential if not fully eliminated during differentiation and manufacturing processes. 49 Although improved differentiation protocols and rigorous quality control measures have substantially reduced this risk, even a small number of contaminating cells could potentially form tumors composed of multiple tissue types after transplantation, necessitating continued efforts to ensure product safety. 50
While gene editing presents a powerful tool to enhance the therapeutic potential of hPSC-β cells, its clinical translation requires careful consideration of safety concerns. First, off-target double-strand breaks caused by Cas9, for instance, can lead to large-scale chromosomal rearrangements or insertional mutations, potentially altering cell identity or increasing tumorigenic risk.51,52 Moreover, integrating viral vectors (e.g. lentiviruses) used for gene delivery can disrupt regulatory elements when inserted near proto-oncogenes such as myelocytomatosis oncogene (MYC) or tumor suppressor 53 (TP53). 53 These insertion events may not be uniformly distributed across the cell population, creating clonal heterogeneity and increasing the probability of malignant transformation within specific subpopulations. In some cases, prolonged in vitro culture of hPSCs can also lead to the acquisition of chromosomal abnormalities characterized by enhanced proliferative capacity and resistance to apoptosis, which confer selective growth advantages and have been recurrently associated with malignant transformation in multiple studies. 54 Additionally, unintended immune activation may occur if engineered hPSC-β cells express immunogenic neoantigens due to aberrant transcription from cryptic promoters, activation of silent viral elements, or mutations arising during gene editing. 55 The combination of risks such as uncontrolled cell proliferation, genomic alterations, and immune-related complications makes it essential to apply strict quality control measures and maintain highly regulated conditions during the development and preparation of hPSC-β cells for clinical use. Furthermore, tracking the fate and behavior of transplanted hPSC-β cells in vivo is critical to assess engraftment success, monitor long-term function, and promptly identify potential safety concerns such as ectopic growth or immune rejection. Therefore, the application of safer and more precise gene editing techniques, as well as comprehensive quality control processes, is essential to ensure the clinical viability of gene-edited hPSC-β cell therapies.
Solutions through gene-edited hPSC-β cell: Enhanced or corrected functionality
Current hPSC-β cells exhibit reduced GSIS compared to primary islets,15,16 prompting efforts to enhance their functional maturity through gene editing strategies (Figure 2). Among the available gene editing systems, Streptococcus pyogenes Cas9 (SpCas9) is the most widely used endonuclease due to its high precision and efficiency. 56 In one representative study, SpCas9 was employed to knock out the SLC30A8 (solute carrier family 30 member 8), which encodes ZnT8, a protein involved in insulin granule maturation and release. By introducing nonsense mutations into the coding region of SLC30A8 in human embryonic stem cells (hESCs), the resulting hESC-derived β cells exhibited significantly enhanced insulin secretion following glucose stimulation. 57 Mechanistically, the knock-out of SLC30A8 relieved zinc-mediated inhibition of insulin release by reducing zinc accumulation within insulin granules, as confirmed by diminished zinquin fluorescence and decreased electron-dense core intensity in transmission electron microscopy (TEM) analysis. Functionally, knock-out of SLC30A8 in hESC-β cells exhibited significantly elevated GSIS (stimulation index (SI): knock-out, 2.23; wild-type, 1.36), increased insulin content per cell, and improved proinsulin-to-insulin processing efficiency (proinsulin/C-peptide ratio reduced to 8.82% vs 13.94% in vivo), leading to superior glycemic control and glucose tolerance when transplanted into streptozotocin (STZ)-induced diabetic mice. That study provided a compelling proof-of-concept that targeted genetic modulation of ion transport pathways can be leveraged to improve the functional maturity and therapeutic efficacy of hESC-β cells for diabetes cell replacement therapy. 57 However, the long-term impact of zinc deficiency on β cell stability remains to be clearly elucidated.

Scheme of recent gene editing strategies for enhancing or correcting insulin secretory function in hPSC-β cells: (a) reduction of intracellular zinc accumulation through knock-out of solute carrier family 30 member 8 (SLC30A8), leading to improved glucose-stimulated insulin secretion (GSIS) and proinsulin processing efficiency, 57 (b) restoration of membrane dynamics for insulin exocytosis via knock-out of zinc finger protein 148 (ZNF148), which facilitates annexin A2 (AnxA2) translocation, 58 and (c) correction of impaired translation initiation and endoplasmic reticulum (ER) processing by repairing the start codon mutation in insulin (INS), thereby restoring insulin production and dynamic secretion both in vitro and in vivo. 60
Next, CRISPR/Cas9-mediated knock-out of zinc finger protein 148 (ZNF148) in hESCs was performed by introducing frameshift mutations into ZNF148 gene. The resulting gene-edited hESC-β cells exhibited substantially enhanced insulin secretion, with static GSIS showing approximately a threefold increase in C-peptide release under high glucose (20 mM) conditions compared to controls. 58 Mechanistically, chromatin immunoprecipitation followed by quantitative polymerase chain reaction (ChIP-qPCR) demonstrated that ZNF148 directly represses the transcription of S100 calcium binding protein A16 (S100A16) that acts as a membrane adaptor. When ZNF148 is deleted, S100A16 expression is upregulated, enabling the formation of a functional complex with annexin A2 (AnxA2). This annexin-S100 complex plays a critical role in reorganizing membrane domains by linking lipids to the actin cytoskeleton. The interaction drives the translocation of AnxA2 from the nucleus to the inner side of the plasma membrane, where it supports membrane remodeling necessary for vesicle docking and insulin granule exocytosis. 59 Thus, this pathway helps restore the secretory function of hESC-β cells by facilitating the final step of insulin release. Functional assays further confirmed significantly improved first- and second-phase secretion kinetics and increased coordinated Ca²⁺ oscillations. These enhancements were recapitulated in primary human islets upon small interfering RNA (siRNA)-mediated knock-down of ZNF148, positioning it as a negative regulator of insulin secretion and a promising target for therapeutic modulation of hESC-β cell functionality. However, the proliferation rate was modestly reduced, which may necessitate adjustment of initial seeding density during large-scale production.
In another example, a homozygous start codon mutation [Insulin (INS)ATG>ATA] in patient-derived human induced pluripotent stem cells (hiPSCs) was corrected with CRISPR/Cas9 and a single-stranded oligonucleotide donor by exploiting the homology-directed repair (HDR) pathway. 60 The gene correction restored insulin biosynthesis and secretion following differentiation into hiPSC-β cells, demonstrating the therapeutic potential of gene editing for monogenic forms of diabetes. The corrected cells exhibited insulin content of 0.6 pmol/cell, comparable to wild-type levels, and secreted 0.027 ± 0.002 pmol insulin following 30.5 mM KCl stimulation, while mutant cells failed to produce or secrete detectable insulin. The start codon mutation at the INS locus disrupts translation initiation, thereby impairing insulin biosynthesis and secretion in hiPSC-β cells. Mechanistically, INS gene correction restored translation initiation, stabilized INS mRNA, and enhanced activation of the unfolded protein response via spliced x-box binding protein 1 (sXBP1), indicating proper endoplasmic reticulum (ER) processing. In vivo, grafting of gene-edited hiPSC-β cell clusters into NSG [non-obese diabetic (NOD) – severe combined immune-deficient (scid) gamma] mice which lack functional T cells, B cells, and NK cells, followed by STZ-induced β cell ablation resulted in sustained normoglycemia, detectable human C-peptide secretion, and dynamic insulin release in response to fasting and re-feeding, confirming functional GSIS restoration. These findings demonstrate that gene correction directly rescues both insulin production and secretion through reactivation of translational competence at the INS locus. Similarly, CRISPR-Cas9 mediated correction of the pathogenic INS or wolfram syndrome 1 (WFS1) mutation in patient-derived hiPSCs led to a substantial enhancement of insulin biosynthesis and secretion following differentiation into hiPSC-β cells.60–64
Building on this strategy, CRISPR-Cas9 was applied to patient-derived hiPSCs carrying a monogenic defect that causes congenital hyperinsulinism (CHI), an infant-onset disorder marked by unregulated insulin secretion. 65 CHI in this patient was traced to a missense mutation (V187D) in the gene of ATP-binding cassette subfamily C member 8 (ABCC8), which encodes the sulfonylurea receptor 1 (SUR1) subunit of the KATP that links glucose metabolism to membrane depolarization and insulin release. 66 The V187D substitution locks the KATP in a closed state, triggering persistent insulin secretion. Using SpCas9 and a single-stranded oligonucleotide donor, the mutant allele was precisely corrected to the wild-type sequence by the HDR pathway. Gene-corrected patient-derived hiPSCs were differentiated into hiPSC-β cells that displayed normal KATP activity and physiologic GSIS, whereas unedited mutant cells retained the hyperinsulinemic phenotype. This proof-of-concept shows that autologous, CRISPR-repaired hiPSC-β cells can rectify single-gene β cell disorders and highlights the feasibility of personalized, gene-edited cell-replacement therapy for genetically defined subtypes of type 1 diabetes (T1D).
Collectively, these studies underscore the therapeutic potential of autologous cell replacement strategies enabled by precise gene editing. As technologies such as base editing and prime editing continue to mature, the correction of pathogenic mutations is becoming increasingly accessible and efficient.67,68 These editing strategies individually target core pillars of β cell functionality such as granule composition, secretory machinery, and protein quality control, and have demonstrated improvements in glucose responsiveness. However, when applied in combination, a systematic analysis is necessary to evaluate potential interactions and their effects on metabolic balance. Despite the personalized advantages of autologous therapies, the complexity of individualized gene editing and manufacturing presents logistical challenges. In contrast, allogeneic cell therapies remain highly attractive due to their off-the-shelf applicability, lower manufacturing costs, and scalability. Consequently, significant efforts are being directed toward the development of gene-edited allogeneic hPSC-β cells, particularly to address the challenge of immune rejection, which will be discussed in the following section.
Solutions through gene-edited hPSC-β cell: Reduced immune rejection
Transplanted allogeneic hPSC-β cells are rapidly rejected by the host immune system through multiple mechanisms, including T cell-mediated cytotoxicity, NK cell recognition, and antibody-mediated responses. 69 This immune rejection typically necessitates systemic immunosuppression, which carries significant clinical risks. To circumvent these challenges, gene editing strategies have been employed to create hypoimmune cell populations capable of evading immune surveillance (Figure 3). CRISPR/Cas9 was used to knock out β2-microglobulin (B2M) and class II major histocompatibility complex transactivator (CIITA), effectively eliminating the expression of MHC class I and II molecules. 70 However, since the loss of MHC class I can trigger NK cell-mediated “missing-self” recognition, CD47, a key immune checkpoint inhibitor, was overexpressed via lentiviral delivery to provide a compensatory “don’t-eat-me” signal. This combination rendered hiPSC-β cells highly immunoevasive. When transplanted intramuscularly into fully immunocompetent rhesus macaques, these hypoimmune hiPSC-β cells survived up to 40 weeks without triggering T cell activation, NK cell cytotoxicity, or antibody-mediated rejection. In contrast, wild-type hiPSC-β cells were rapidly rejected within 1 week, with complete graft clearance by week 2 and no observed glycemic improvement due to early cell loss. Mechanistically, CD47 inhibited innate immune activation by engaging signal regulatory protein α (SIRPα), while MHC deletion abrogated recognition by adaptive T cells. However, more recent findings suggest that CD47 overexpression may suppress insulin secretion in hESC-β cells, highlighting the need to validate whether immune evasion compromises β cell function. 71

Schematic summary of recent gene editing strategies to overcome alloimmune and autoimmune rejection: (a) major histocompatibility complex (MHC) class I/II deletion [β2-microglobulin (B2M)⁻/⁻, class II major histocompatibility complex transactivator (CIITA)⁻/⁻] and CD47 overexpression to suppress both adaptive and innate immune responses. 70 ; knock-in of immunomodulatory cytokines, mutant interleukin-2 (IL-2 N88D), interleukin-10 (IL-10), and transforming growth factor-β (TGF-β) to induce local immune tolerance, 72 (b) Renalase (RNLS) knock-out to reduce endoplasmic reticulum (ER) stress-induced antigen presentation and T cell activation, 75 and (c) C-X-C motif chemokine ligand 10 (CXCL10) knock-out to disrupt early chemokine-mediated cytotoxic immune cell recruitment. 76
To induce immune tolerance, hiPSC-β cells were engineered with B2M knock-out to remove HLA class I expression, and knock-in of immunomodulatory cytokines such as interleukin-10 (IL-10), transforming growth factor-β (TGF-β), and a mutant interleukin-2 (IL-2 N88D) at the glyceraldehyde 3-phosphate dehydrogenase (GAPDH) locus. 72 The gene-edited hiPSC-β cells maintained approximately 30% co-expression of NKX6.1 and C-peptide, demonstrated normal GSIS in vitro, and survived for at least 8 weeks when transplanted into autoimmune NOD mice, which exhibit impaired functional regulatory T cells, without evidence of rejection, while wild-type hiPSC-β cells were rejected within 2 weeks and failed to restore normoglycemia. This dual strategy reduced both innate and adaptive immune responses by creating a tolerogenic microenvironment and eliminating direct recognition by host T and NK cells. However, the constitutive expression of immunomodulatory cytokines may impair local immune surveillance, potentially increasing the risk of undetected infections at the graft site.
A subsequent strategy advanced the immune-evasion design by combining CRISPR/Cas9-mediated knock-outs of MHC class I and II (via B2M and CIITA), overexpression of CD47, and additional expression of CD64, a high-affinity Fc receptor that binds circulating immunoglobulin G (IgG). 73 As a result, these gene-edited hiPSC-β cells were resistant to both antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC), even when exposed to clinically relevant concentrations of anti-HLA and non-HLA antibodies such as anti-MHC class I polypeptide-related sequence A (MICA) and anti-CD52. In vivo, these gene-edited hiPSC-β cells survived for at least 4 weeks after subcutaneous transplantation into NSG mice, co-injected with human NK cells and alemtuzumab, while wild-type hiPSC-β cells were eliminated within 1 week and failed to form detectable grafts. The CD64 receptor functioned by sequestering the Fc region of IgG, thereby preventing activation of downstream immune effector pathways. This Fc-capturing mechanism, when combined with classical hypoimmune edits, allowed for full resistance to both cellular and humoral rejection; however, sustained high-affinity binding and internalization of IgG immune complexes may impose intracellular stress, potentially overloading lysosomal degradation pathways and compromising long-term cellular homeostasis. Although these strategies show strong immune resistance, potential drawbacks such as intracellular stress from lysosomal overload and impaired immune surveillance must be carefully addressed to ensure long-term safety and clinical viability.
A different strategy employed stepwise CRISPR/Cas9 editing to delete highly polymorphic HLA-A, HLA-B, and HLA-C genes, along with CIITA, while selectively retaining HLA-A2 expression. 74 These hiPSC-β cells successfully differentiated into insulin-producing cells without compromising lineage identity or GSIS functionality. Importantly, they exhibited reduced NK cell activation in vitro and survived for at least 20 days when transplanted into humanized NSG mice reconstituted with HLA-A2-positive peripheral blood mononuclear cells (PBMCs), while wild-type hiPSC-β cells were rejected within 2 weeks and failed to maintain graft integrity or functional marker expression. The retained HLA-A2 expression preserved HLA-E levels, which in turn inhibited NK cells through an inhibitory NK cell receptor, thereby preventing the “missing-self” response while still minimizing T cell recognition. This immune engineering strategy reduces both innate and adaptive immune rejection while maintaining immune surveillance, thus establishing a promising platform for allogeneic hiPSC-β cell transplantation without the need for long-term immunosuppression. However, its reliance on partial HLA matching may limit donor–recipient compatibility across diverse populations and complicate universal applicability.
Separately, β cell-intrinsic factors influencing immune-mediated destruction have also been explored. A genome-wide in vivo CRISPR screen in NOD mice identified renalase (RNLS) as a critical modulator of β cell susceptibility. 75 Knocking out RNLS in hiPSC-β cells enhanced resistance to apoptosis under ER stress, suggesting that RNLS contributes to β cell vulnerability by modulating stress response pathways. This protective effect was associated with attenuated activation of the unfolded protein response and reduced expression of pro-apoptotic factors. Functionally, RNLS-deficient hiPSC-β cells survived for over 2 months after transplantation into diabetic NOD mice, whereas wild-type hiPSC-β cells were eliminated within 1–2 weeks and failed to sustain insulin secretion or glycemic control. From an immunological standpoint, this gene editing approach enhances the resilience of hiPSC-β cells to autoimmune attack without compromising allo-recognition or proliferative control, thereby offering a refined strategy for clinically viable and immune-compatible β cell replacement in T1D. However, despite these benefits, the knock-out of RNLS may inadvertently impair graft–host communication required for long-term integration and regeneration, given its role in immunometabolic signaling and tissue repair.
Lastly, a comprehensive CRISPR screen combined with single-cell RNA sequencing was used to identify cytokines critical for immune rejection. 76 In this study, CXCL10, induced by IFN-γ, was identified as a key factor in recruiting cytotoxic immune cells. Deletion of CXCL10 in hESC-β cells led to a twofold increase in cell survival when co-cultured with PBMCs, T cells, or NK cells in vitro. Furthermore, in NSG mice lacking T, B, and NK cells, both wild-type and CXCL10-deficient hESC-β cells initially survived and maintained insulin secretion. However, upon adoptive transfer of human PBMCs, only the CXCL10-deficient hESC-β cells retained function up to at least 17 weeks, whereas wild-type hESC-β cells lost insulin secretion capacity by week 11, indicating enhanced resistance to immune-mediated rejection. Mechanistically, CXCL10 knock-out suppressed early chemokine-mediated immune cell infiltration via the CXC chemokine receptor 3 (CXCR3) signaling pathway. These findings demonstrate that precise targeting of early inflammatory signaling pathways in hESC-β cells, specifically the IFN–CXCL10–CXCR3 axis, offers a potent strategy to mitigate alloimmune rejection and improve graft survival without systemic immunosuppression.
In a related approach, CRISPR was used to knock out B2M and incorporate an inducible system for programmed death-ligand 1 (PD-L1) expression. This dual modification protected the grafts from diabetogenic CD8⁺ T cell–mediated destruction following adoptive transfer of human PBMCs into NSG mice, whereas wild-type hESC-β cells were rapidly rejected and failed to maintain insulin expression. 77 Nevertheless, sustained suppression of CXCL10-mediated chemotaxis and prolonged PD-L1-driven checkpoint signaling may dampen graft–host immunological cross-talk essential for vascularization and pathogen surveillance, potentially delaying tissue repair and increasing susceptibility to opportunistic infection or tumor immune evasion.
Overall, the hypoimmune engineering strategy is a promising approach that enables the widespread use of hPSC-β cells through off-the-shelf production. This strategy is gaining significant attention not only for β cell therapies but also for the development of various stem cell-derived cell therapies. However, concerns regarding biosafety arise due to the immune evasion capabilities of these cells. For example, they may evade elimination by the immune system when they become cancerous. Therefore, multilayered immune-evasion strategies must be carefully combined in a stepwise manner to offset individual risks, and long-term monitoring is essential to assess the potential for tumorigenesis or systemic immune suppression.
Solutions through gene-edited hPSC-β cell: Enhanced biosafety
To minimize the risk of teratoma formation and unwanted mutations arising from residual undifferentiated cells, recent strategies have incorporated lineage-specific reporter systems to selectively enrich for functionally committed hPSC-β cells, thereby enhancing the overall biosafety and therapeutic precision of the cell product (Figure 4). In one approach, CRISPR/Cas9-mediated homologous recombination was used to generate hiPSC-β cells with a precise knock-in of enhanced green fluorescent protein (EGFP) at the C-terminus of the endogenous PDX1 locus, enabling real-time monitoring of β cell lineage commitment during differentiation. 78 In this study, the off-target analysis of 16 high-risk genomic loci showed no unintended mutations, indicating high genome integrity. The gene-edited hiPSC-β cells maintained a normal karyotype (46, XY), expressed pluripotency markers [OCT4, SOX2, stage-specific embryonic antigen-4 (SSEA4), tumor rejection antigen-1-60/-81 (TRA-1-60/-81)] and retained trilineage differentiation potential with clear β cell differentiation as evidenced by co-expression of insulin and EGFP in vitro. Functionally, the reporter system enabled non-invasive tracking of insulin-producing cells post-differentiation, reducing the need for destructive assays and enhancing the monitoring precision of β cell lineage commitment. From a biosafety perspective, this strategy allows for precise lineage verification, minimizes the risk of misidentification or contamination, and provides a platform for eliminating residual undifferentiated cells, thereby enhancing the safety of hiPSC-β cell based therapeutic applications. However, the integration of exogenous reporter genes may introduce the risk of insertional mutagenesis, making thorough characterization of the insertion site essential to ensure genomic stability.

Schematic summary of recent gene editing strategies to improve safety and traceability: (a) Knock-in of pancreatic and duodenal homeobox 1 (PDX1)-enhanced green fluorescent protein (EGFP) for real-time monitoring of lineage commitment and elimination of off-target or undifferentiated cells, 78 (b) knock-in of insulin (INS)–green fluorescent protein (GFP) for in vivo tracking of hPSC-β cells or islet-like organoids and assessment of graft purity, 79 and (c) marker-free gene insertion using CRISPR/Cas9 ribonucleoproteins (RNPs) and adeno-associated virus serotype 6 (AAV6) vectors for precise integration of therapeutic genes while minimizing long-term immunogenic and oncogenic risks. 82
A CRISPR/Cas9-based knock-in of a green fluorescent protein (GFP) reporter at the endogenous INS locus was also used to generate human islet-like organoids (HILOs) with traceable hiPSC-β cell identity, enabling non-invasive monitoring of insulin-producing cells throughout differentiation and post-transplantation. 79 This reporter system provided a robust platform to confirm β cell-lineage fidelity, detect off-target endocrine populations, and track graft localization in vivo using fluorescence imaging. From a biosafety perspective, INS-GFP knock-in allows real-time assessment of differentiation efficiency and purity, which is critical for minimizing the risk of undifferentiated residual cells or polyhormonal contaminants, both of which are associated with tumorigenicity and graft failure. Combined with immune-evasive PD-L1 engineering, this strategy supports safe, trackable, and durable hiPSC-β cell therapy by providing both functional immune protection and rigorous identity validation.
Many of the aforementioned gene editing strategies introduced selection markers such as antibiotic-resistance genes (e.g. puromycin or neomycin resistance cassettes) or fluorescent reporters into hPSCs to isolate gene-edited populations. However, the permanent integration of such markers poses biosafety concerns due to their potential immunogenicity or oncogenicity. To address this, marker-free gene editing technologies have been developed to achieve precise genome modifications without incorporating exogenous selection cassettes.80,81 This approach typically involves the use of transient delivery systems, such as Cas9 ribonucleoproteins (RNPs) and non-integrating donor templates, to enable efficient HDR pathway without leaving behind exogenous genetic elements. In a recent study, CRISPR/Cas9 RNPs were combined with adeno-associated virus serotype 6 (AAV6) donor templates, an episomal and generally non-integrating vector system, to achieve homology-directed knock-in efficiencies of up to 94% at multiple genomic loci in hESCs, while fully avoiding the integration of potentially oncogenic or immunogenic selection cassettes typically associated with lentiviral systems. 82 These findings underscore the translational potential of marker-free editing platforms for generating safer and more precisely engineered stem cell therapies. Separately, edited clones preserved normal karyotypes and pluripotency markers, and deep sequencing confirmed that high-fidelity Cas9 minimized off-target mutations; critically, no TP53 variants or chromosomal anomalies were detected, further supporting the safety of this editing paradigm. 82 This marker-free gene editing strategy offers a safer and clinically applicable route for generating gene-corrected stem cell therapies. However, whole-genome sequencing remains critical to detect rare mutations, particularly in noncoding regulatory regions or tumor suppressor genes, that could compromise long-term safety.
Above strategies facilitate cell selection during the manufacturing process and enable post-transplantation tracking. In parallel, the use of safety switches offers a means to address biosafety concerns associated with stem cell-derived therapeutics by allowing for the rapid elimination of cells in the event of adverse outcomes such as tumorigenesis. In fact, similar approaches have been actively pursued in the context of chimeric antigen receptor (CAR)-T cell therapy, where safety switches are introduced to enable prompt clearance of the therapeutic cells if necessary.50,83–85 These strategies are now being increasingly applied to stem cell-derived tissues, and their successful implementation in such systems suggests their potential applicability to therapeutic hPSC-β cells to enhance safety. Ultimately, biological safety is evolving toward a multilayered framework in which independent, orthogonal safeguards are combined to preserve therapeutic efficacy while ensuring traceability and removability of the transplanted cells.
Current clinical progress of gene-edited hPSC-β cell
While gene editing mitigates immunogenicity at the cellular level, tissue engineering strategies like encapsulation have been actively explored to provide additional physical protection against immune attack. By physically isolating transplanted hPSC-β cells from host immune effectors, encapsulation supports localized immune evasion and can extend graft survival without the need for systemic immunosuppression.86,87 The convergence of gene editing with encapsulation technologies may facilitate development of next-generation hPSC-β cell therapies with enhanced functional durability and immune protection. This integrative strategy could help overcome major translational barriers and potentially accelerate the clinical realization of hPSC-β cell replacement therapies for T1D.
Recent clinical trials have begun evaluating gene-edited stem cell-derived PECs for the treatment of T1D. For instance, clinical trials NCT05210530 and NCT05565248 are currently assessing the safety and efficacy of such cell products in human subjects. 88 In trial NCT05210530, the investigational product VCTX210A consists of hESC-derived PECs (PEC210A) that were engineered with CRISPR-mediated gene editing. The genetic modifications include knock-out of B2M to eliminate MHC class I expression, along with overexpression of PD-L1 and HLA-E, both of which contribute to immune evasion by suppressing cytotoxic T cell and NK cell activity. Additionally, TXNIP was knocked out to protect β cells from oxidative and ER stresses. These gene-edited cells are delivered using the PEC-Direct macroencapsulation device (MED), which features a perforated membrane that allows vascular infiltration and facilitates nutrient exchange while exposing the cells to the host immune environment. The phase 1 trial NCT05210530 began in January 2022 and was completed in early 2023, enrolling 10 T1D patients. Although the results have not yet been posted, the trial established initial safety and feasibility in humans.
Thereafter, NCT05565248, a phase 1/2 trial investigating the next-generation product VCTX211 was initiated in January 2023 and is actively recruiting up to 40 participants, with an estimated primary completion date of August 31, 2025. Notably, both trials employ gene-edited PECs, rather than fully mature hPSC-β cells, at the transplantation stage. This reflects the persistent challenge of achieving complete in vivo maturation and highlights the need for gene editing strategies that directly enhance hPSC-β cell functional development post-transplantation. Trial NCT05565248 investigates an advanced platform, VCTX211, which utilizes the same PEC-Direct MED and builds upon the PEC210A cell line by incorporating additional gene edits. In VCTX211, the PECs also overexpress A20 (encoded by tumor necrosis factor alpha-induced protein 3, TNFAIP3), a ubiquitin-modifying enzyme that inhibits nuclear factor-κB (NF-κB)-mediated inflammatory signaling, and mesencephalic astrocyte-derived neurotrophic factor (MANF), which promotes β cell survival under metabolic and inflammatory stress. The encapsulation strategy is designed to support revascularization within the implant while minimizing the requirement for systemic immunosuppressants. These trials are expected to demonstrate whether gene-edited hPSC-β cell therapies can achieve long-term insulin secretion and glycemic regulation in patients with T1D by combining immune-evasive cellular engineering with device-enabled local vascular integration.
Independent of gene editing approaches, the VX-880 program by Vertex Pharmaceuticals exemplifies the most clinically advanced application of hESC-β cell replacement therapy for T1D to date. Initiated as a phase 1 trial (NCT04786262) in 2021, VX-880 (also known as zimislecel) demonstrated promising safety and efficacy signals, including restored C-peptide production and reduced insulin dependence in patients receiving partial islet cell infusions under immunosuppression. 89 These results supported its advancement into a phase 3 clinical trial, which began in 2025, where full-dose administration aims to confirm durable glycemic control, elimination of severe hypoglycemic episodes, and significant reductions in hemoglobin A1c levels. According to a May 2025 update, VX-880 has progressed into a phase 3 clinical trial, marking a historic milestone as the first hESC-β cell therapy to reach this stage. This advancement establishes the clinical feasibility of stem cell–based replacement therapy for T1D.
Conclusion and outlook
The advancement of gene-edited hPSC-β cells represents a significant milestone in the pursuit of an effective and durable treatment for T1D. By enabling sustained and physiologically regulated insulin secretion, this approach offers the potential to reduce or eliminate dependence on exogenous insulin. Importantly, when combined with optimized differentiation protocols and advanced delivery platforms, gene editing serves as a powerful tool to address key barriers such as functional immaturity, immune rejection, and biosafety for hPSC-β cell–based therapy. As these technologies move closer to clinical application, continued efforts toward improving precision, scalability, and safety through refined editing tools and rigorous quality control will be essential to ensure therapeutic efficacy and long-term success.
A primary challenge impeding the clinical translation of hPSC-β cells is their incomplete functional maturation, which limits their ability to recapitulate the tightly regulated glucose responsiveness of native pancreatic β cells. Recent efforts have focused on refining stepwise differentiation protocols and introducing lineage-specific transcription factors to enhance β cell functionality. In parallel, gene editing strategies targeting key regulators of insulin secretion, metabolic coupling, and cellular stress responses have shown promise in improving the therapeutic performance of hPSC-β cells. Another key area requiring further investigation is the metabolic efficiency of hPSC-β cells, as ATP availability critically regulates GSIS. Given the high energy demands of β cells, enhancing mitochondrial function through strategies such as promoting oxidative phosphorylation, modulating glycolytic flux, or introducing metabolic regulators may improve both insulin secretion and post-transplant cell survival.
Beyond functional immaturity, immune rejection remains a critical hurdle for successful hPSC-β cell transplantation. While encapsulation technologies have been extensively explored as a tissue engineering approach to provide physical separation from host immune components, they may face limitations such as pericapsular fibrosis and restricted nutrient exchange. To complement these strategies, immune-evasive engineering of hPSC-β cells through advanced gene editing such as targeted deletion of MHC or overexpression of immune checkpoint proteins offers an additional layer of protection. The synergistic use of tissue engineering and genetic strategies may thus provide a more robust and durable solution to overcoming immune barriers in β cell replacement therapy. Such modifications can enable hPSC-β cells to evade immune detection without necessitating lifelong immunosuppressive therapy, thus improving their viability and therapeutic potential. However, the clinical application of hypoimmune hPSC-β cells will require parallel advances in safety technologies, as complete immune evasion can allow malignant or aberrant cells to persist undetected. Accordingly, the development of robust biosafety switches and rigorous monitoring systems will be essential to ensure the safe and practical realization of hypoimmune hPSC-β cell therapies.
To enhance both safety and efficacy, future strategies may combine gene-edited hPSC-β cells with advanced tissue engineering platforms, non-viral gene delivery systems, or their combination. Among non-viral approaches, lipid nanoparticle (LNP)-based systems may offer a non-integrating, clinically scalable method for in vivo gene modulation. A recent study has demonstrated that LNPs can deliver mRNA to pancreatic β cells via macrophage-mediated transfer, highlighting a promising route for β cell–specific gene delivery. 90 In parallel, biodegradable scaffolds and immunomodulatory hydrogels can facilitate localized engraftment, vascular integration, and protection from immune attack.87,91–93 By integrating these technologies, it may be possible to improve not only therapeutic durability and immune compatibility but also streamline regulatory approval by reducing reliance on viral vectors.94,95 These integrated platforms may ultimately offer a dynamic microenvironment that improves β cell maturation, immune protection, and controllable gene modulation after transplantation, although further research is needed to validate their clinical utility.
In summary, gene-edited hPSC-β cells represent a major step forward in T1D therapy, offering the potential for durable insulin independence. Ongoing efforts to improve their functionality such as metabolic performance, immune evasion, and safety will be essential to realize their full clinical potential. With continued innovation, stem cell-derived β cell therapy may not only transform diabetes care but also advance the broader field of regenerative medicine.
Footnotes
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was supported by the Korean Fund for Regenerative Medicine (KFRM) grant funded by the Korean government (Code: KFRM24A0105L1). This work was also supported by the Incheon National University Research Grant in 2022.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.