
Abstract
Spiral ganglia neurons (SGNs) impairment can cause deafness. One important therapeutic approach involves utilizing stem cells to restore impaired auditory circuitry. Nevertheless, the inadequate implementation of research methodologies poses a challenge in accurately assessing the functionality of derived cells within the circuit. Here, we describe a novel method for converting human embryonic stem cells (hESCs) into otic neurons (ONs) and assess their functional connectivity using an optogenetic approach with cells or an organotypic slice of rat cochlear nucleus (CN) in coculture. Embryonic stem cell-derived otic neurons (eONs) exhibited SGN marker expression and generated functional synaptic connection when cocultured with cochlear nucleus neurons (CNNs). Synapsin 1 and VGLUT expression are found in the cochlear nucleus of brain slices, where eONs projected processes during the coculture of eONs and CN brain slices. Action potential spikes and INa+/IK+ of CNNs increased in tandem with light stimulations to eONs. These findings provide further evidence that eONs may be a candidate source to treat SGN-deafness.

Introduction
It was predicted that about one-quarter of the global population will suffer from hearing loss diseases by 2050. 1 Hearing loss influences people’s lives extensively, including communication, cognition, education, and even mental health, posing a socioeconomic challenge.1–3 The most prevalent form of hearing loss disease is sensorineural hearing loss (SNHL), which accounts for the majority of cases.4,5
The inner ear’s spiral ganglion neurons (SGNs) and hair cells work together to send sound impulses to the brainstem.6,7 The external ear detects sound, and hair cells are in charge of translating mechanical signals into chemical and electrical signals so that they can travel downstream via oscillations. 8 The SGN is a typical bipolar neuron, which is a kind of adult cell lacking regenerative ability. 9 The spiral ganglion is responsible for transmitting peripheral sound information to the cochlear nucleus in the brainstem, and then to the auditory cortex.10,11
Hair cell damage-related hearing impairments can be treated with cochlear implants to restore hearing,12,13 however, degeneration or loss of SGNs can result in irreversible hearing loss because they lack the capacity to regenerate. 10 Roughly 10% of individuals with hearing impairment are thought to have SNHL. 14 While the influence of hair cell loss can be partially mitigated by cochlear implantation, the loss of auditory neurons cannot be treated conventionally as the expected performance of implantation is limited by poor innervation and the quantity of remaining SGNs.15,16 Thus, it’s critical to develop potential strategies, such as stem cell replacement therapy, to lessen the damage caused by SGNs.
Stem cell research has advanced the treatment of auditory neuropathy illnesses by using stem cells to replace degraded SGNs. Apart from the investigation of neural stem cells (NSCs) derived from non-pluripotent stem cells (PSCs), which include human fetal inner ear, 17 cochlear Sox2+ glial Cells, 18 brain, 19 olfactory, 20 and mesenchymal stem cells, 21 pluripotent stem cells (PSCs), which include mouse embryonic stem cells (ESCs), 22 human ESC (hESC)-derived otic progenitors, 23 neurosensory-like progenitors induced from human induced pluripotent stem cells (hiPSCs), 24 and otic sensory neurons generated from mESCs in 3D culture, 25 were also studied for different purposes such as cell therapy and modeling.
More signals involved in the development of SGN are used to induce putative auditory neurons as the understanding of developmental biology advances. Using BMP4 induction, 26 FGF3 and FGF10 induction followed by mechanical purification of otic neural progenitors (ONPs), 23 simulating the SGN developmental process followed by FACS purification of ONPs, 27 and organoid culture in 3D, 25 we and others have induced mouse or human ESCs to differentiate into cells with SGN-like characteristics.
ONPs generated from PSCs have been injected into the inner ear, in addition to in vitro research, to monitor the fate and behavior of the cells.22,23,28–31 We showed that transplanting hESC-derived otic progenitors into an auditory neuropathy model could exhibit the restoration of auditory evoked responses. 23 However, further research is required to determine whether these cells could integrate functionally into the auditory circuit.
To determine whether synapses and electrophysiological activity could be observed between mESC-derived SGN-like cells and neurons derived from the cochlear nucleus (CN), a cellular coculture system was employed. 32 Organotypic cocultures of CN brain slices and hPSC-derived neurons were also established to validate synapse formation and neuron innervation. 33 However, how to functionally integrate stem cell-derived neurons into the native nervous system remains elusive.
To confirm the functional connection between various parts of neural circuitry, optogenetic techniques can offer a solution by pre-labeling cells with light-sensitive ion channels to selectively stimulate a particular group of cells.34–38 Nevertheless, the potential circuitry between exogenous neurons and natural CN or cochlear nucleus neurons (CNNs) has not been assessed using this method.
Here, we provide an alternative and more straightforward induction protocol recapitulating ONP development to generate auditory neurons from hESCs that have undergone optogenetic modification. eONPs were purified with differential selection by collagenase. The SGN-like properties of eONs were demonstrated by immunocytochemistry, qPCR, and electrophysiology data. The establishment of functional synapses and functional integration between eONs and CNNs in cells and at the organotypic level following coculture was validated by immunostaining and optogenetics results. Our research shows that SGN-like cells derived from hESCs have a lot of clinical potential and could be useful in creating a novel therapeutic approach to treat SNHL-related disorders.
Experimental procedures
Animals
Postnatal Sprague-Dawley (SD) rat pups (P12–P14, n = 10) used for organotypic brain slice (BS) culture were obtained from Hangzhou Medical College. The animals were kept in standard conditions, with food and water available at all times. All experiment procedures were approved by the Hangzhou Medical College and the Ethics Committee.
Construction of optogenetic ESC line and EGFP-hESCs line
The hESC H1 and H9 cells were obtained from Stem Cell Bank of Chinese Academy of Sciences. To facilitate detailed visualization of cellular behavior and enhance the study of neuronal differentiation, we established the hESC-H9-EGFP line. This cell line was established through the stable integration of an EGFP-expressing plasmid (Supplemental Figure 1A, Addgene) into the H9 hESC genome via electroporation using the CUY21 system (BEX CO., LTD.). For purposes of optogenetic manipulation, we further modified the H9 cell line by transducing it with a lentiviral vector carrying the hChR2(H134R)-eYFP construct (Addgene, Supplemental Figure 1B). This construct comprises the light-sensitive channelrhodopsin-2 (ChR2) variant H134R, fused to enhanced yellow fluorescent protein (eYFP), which facilitates both optical stimulation and visualization of the transduced cells. The lentiviral packaging was accomplished using the helper plasmids pCMVR8.74 and psPAX2 (Addgene). In comparative assessments, the H9-EGFP line demonstrated a more robust fluorescence signal than the H9-ChR2-eYFP line, providing enhanced capabilities for tracking cell migration, monitoring neurite outgrowth, and conducting colocalization studies with neurospecific markers.
For stable hESC line establishment with fluorescence, following electroporation or lentiviral cell transduction, single-cell dissociation with Accutase (GIBCO) was performed. The dissociated cells were transferred onto the mouse feeders to allow clone formation in the medium which consists of Knockout™ DMEM (GIBCO), 20% KSR (GIBCO), 1% penicillin/streptomycin (Fdbio science), 1% Non-Essential Amino Acids (GIBCO), 1% GlutaMAX (GIBCO), 100 μM β-Mercaptoethanol (Invitrogen), 8 ng/mL bFGF (Peprotech). Fluorescent cells expressing EGFP/eYFP were selectively grown based on the clonogenic properties of hESCs, resulting in the development of clonal populations. Clones displaying bright and uniform fluorescence were identified under a microscope and subsequently transferred to Matrigel (Corning)-coated culture flasks without feeders for expansion in mTeSR1 medium (STEMCELL Technologies) with daily media changes. The selected EGFP/eYFP-expressing clone was designated as passage 0 (P0). Subsequent passaging was performed at a 1:5 ratio using Accutase (GIBCO). For our experiments, we utilized hESCs between passages P20 and P30 to ensure stable transgene expression while minimizing the risk of accumulated mutations. Cells were cryopreserved in liquid nitrogen using GMP-grade Protein-Free Cryopreservation Medium (Meilunbio) and rapidly thawed in a 37°C water bath for resuscitation prior to experimental use.
The hESC H1 cell line was also utilized for induction, proliferation, and differentiation into eONs.
Induction of eONPs stepwisely from hESCs
To promote the induction of hESCs toward otic neural progenitors (ONPs), a stepwise protocol with specific growth factors and small molecule inhibitors was employed. Initially, hESCs were passaged using Accutase and subsequently cultured in mTeSR1 medium for 72 h to ensure cellular readiness for induction. Following this preparation phase, the culture medium was transitioned from mTeSR1 to Basic Growth Medium (BGM), which is a chemically defined formulation consisting of DMEM/F12 GlutaMAX (GIBCO) supplemented with 2% Albumin, Bovine Serum, Fraction V, Fatty Acid-Poor, Endotoxin-Free (BSA, Millipore), 10 ng/mL Heregulin β-1 (Peprotech), 10 μg/mL Bovine transferrin (Holo form, Invitrogen), 200 ng/mL Animal-Free Recombinant Human IGF-I LR3 (Peprotech), 50 μg/mL (+)-Sodium l-ascorbate (Invitrogen), 0.1% Trace elements A/B/C (Cellgro), 1% Non-Essential Amino Acids, 8 ng/mL bFGF, 1% penicillin/streptomycin, 100 μM β-Mercaptoethanol, and 20 μM SB431542 (Selleck).
In accordance with the principles of dual SMAD inhibition, the addition of the small molecule inhibitors LDN193189 2HCl and IWP-2, alongside SB431542, initiated the induction of hESCs. The cells were cultured in BGM for 13 days, with LDN193189 2HCl (500 nM, Peprotech) administered during the initial 3 days. Inhibition of Wnt signaling was achieved by incorporating IWP-2 (2 μM, Peprotech) into the medium for the first 5 days to steer cell differentiation toward a pre-placodal fate. Daily media changes were implemented from day 0 to 13.
Commencing on day 7, the protocol incorporated fibroblast growth factors (FGFs) to support otic lineage specification and proliferation. Specifically, recombinant human FGF3 (rhFGF3) and rhFGF10 (both sourced from R&D Systems) were supplemented at a concentration of 50 ng/mL each into the BGM. Subsequent media changes were performed bi-daily.
Since day 13, the culture medium was replaced with a defined DFNB medium, composed of DMEM/F12 GlutaMAX supplemented with 1% penicillin/streptomycin, 1% N2 supplement (GIBCO), and 2% B27 supplement (GIBCO). Both rhFGF3 and rhFGF10 continued at a concentration of 50 ng/mL each, maintained until day 19 with medium changes every 2 days. This defined culture system facilitated a controlled and efficient induction of hESCs into ONPs.
Purification of eONPs
Following a 19-day induction period of hESCs, the cells were treated with 1 mg/mL collagenase IV (Sigma) at 37℃ to facilitate the isolation of high-purity eONPs from the heterogeneous cell population. Leveraging the differential sensitivities of various cell types to collagenase IV, this enzyme-mediated dissociation allowed for the selective separation of eONPs from non-ONP cells. Specifically, non-ONP cells, exhibiting greater susceptibility to collagenase IV, were preferentially digested and subsequently removed from the culture. In contrast, eONPs, which demonstrated insensitivity to this initial enzymatic treatment, were retained within the culture flask. These remaining eONPs were then subjected to subsequent digestion using Accutase to prepare them for further applications, including passaging, proliferation, directed differentiation, or incorporation into coculture systems.
Proliferation and differentiation of eONPs into eONs
Following the purification of the eONPs, they were induced into differentiated eONs either before or after the proliferation stage. During the proliferation phase, they can be expanded in DFNB medium supplemented with both FGF3 and FGF10 for a duration of 2–3 months. During the differentiation phase, the differentiation medium (DM) used consisted of DFNB medium with the addition of 10 ng/mL BDNF (Peprotech), 10 ng/mL NT3 (Peprotech), 10 ng/mL β-NGF (Peprotech), 10 ng/mL GDNF (Peprotech), 0.5 mM Dibutyryl-cAMP (Sigma), 200 μM L-ascorbate, and 1% penicillin/streptomycin. The precise duration needed for the differentiation of eONs is determined based on the observation of morphological alterations toward neuronal cells. Typically, a continuous culture period lasting 1–2 weeks is conducted, with the differentiation medium being replaced every other day.
Acquisition of CNNs and cocultured with eONs
The cochlear nuclei of 12-day-old Sprague-Dawley rats were dissected using surgical instruments and divided into small tissue blocks. Following digestion using Accutase, the cochlear nuclei cells were incubated in a controlled environment at a temperature of 37°C and a CO2 concentration of 5%. After adhesion, cells of the cochlear nuclei were cocultured with eONs-EGFP or eONs-ChR2-eYFP for subsequent immunofluorescence or optogenetic electrophysiological detection, correspondingly. The culture medium of CNNs, and cocultured cells was composed of DFNB medium, supplemented with 10 ng/mL IGF-1, 10 ng/mL bFGF (Peprotech), and 10 ng/mL EGF (Peprotech).
Acquisition of brain slices and coculture of eON spheres with cochlear nucleus of brain slices (CN-BS)
We established a coculture system consisting of otic neural spheres and rat brainstem slices to investigate their mutual interaction. Initially, eONs were enzymatically dissociated from the culture dishes and subsequently transferred into ultra-low adsorption cell culture plates. Within 1–2 days of suspension culture, the individual eONs coalesced into spherical aggregates. These otic neural spheres, characterized by their uniform diameter of approximately 100 μm, served as the foundational units for subsequent coculture experiments with rat brainstem slices. Following the administration of anesthesia to the SD rat, the rat’s brain was promptly removed and immersed in a cutting solution known as artificial cerebrospinal fluid (ACSF). The ACSF slicing solution was prepared in advance into an ice-water mixture state, and a continuous supply of 95% oxygen and 5% carbon dioxide gas was supplied to the liquid. After rapid dissection of the brain tissue, the excess brain region was removed, and the brainstem containing the cochlear nucleus was affixed to the cutting table using adhesive. The brain slices, which were 200 μm thick and sliced using a Leica VT 1200s, were placed onto the Matrigel-coated insert using a brush. The otic neural spheres of EGFP or ChR2-eYFP were transplanted adjacent to the AVCN region of the brain slice on the following day in the coculture medium. The coculture medium was changed daily. The medium consisted of DMEM high glucose (GIBCO), 5% horse serum (Beyotime Biotechnology), 10% FBS (GIBCO), 1% penicillin/streptomycin/amphotericin B (Beyotime Biotechnology), 25 mM HEPES (GIBCO), 1% N2 supplement, 2% B27 supplement, and 10 ng/mL β-NGF (Peprotech). The immunofluorescence and electrophysiological evaluation were conducted after a period of 10 days of coculture.
Electrophysiological assessments
During the recording process, the glass coverslip containing cells is transferred to the electrophysiological recording chamber, with the recording liquid temperature maintained at 28 ± 2°C. Neurons with smooth surfaces and clear outlines are selected for whole-cell recordings under a 40× Nikon upright microscope. The recording solution used is ACSF, continuously perfused with a gas mixture. Firstly, for recording spontaneous excitatory postsynaptic currents (sEPSC), a cesium methanesulfonate electrode internal solution is employed, with the electrode impedance set at 4–7 MΩ. After membrane rupture, cells with an access resistance (Ra) less than 20 MΩ are statistically recorded. The holding potential is clamped at −60 mV during sEPSC recording. Secondly, when recording neuronal action potentials (AP), a potassium gluconate electrode internal solution is used, and the electrode impedance is set at 4–7 MΩ. Cells are voltage-clamped, and the membrane is ruptured when the resistance exceeds 1 GΩ, followed by switching to current-clamp mode for AP recording. The protocol is as follows: starting injection current at −50 pA, recording time of 500 ms, with increments of 10 pA for 10 sweeps to observe action potential firing. Resting membrane potential is indicated by the voltage at the current baseline during AP recording; action potential threshold is the minimum voltage for action potential initiation; input resistance and time constant (τ) are fitted using Clampfit software; action potential amplitude is measured from threshold to peak; action potential duration is taken as the time course of half-peak amplitude. Thirdly, recording potassium (K+) currents is done using a potassium gluconate electrode internal solution, with the electrode impedance of approximately 4–7 MΩ. In voltage-clamp mode, cells are patched, and potassium currents are recorded after membrane rupture. The protocol is set as follows: starting injection voltage at 0 mV, recording time of 500 ms, with increments of 10 mV for 10 sweeps up to +50 mV to observe potassium ion current emission. When recording sodium (Na+) currents with the CsCl electrode internal solution, the protocol is set as follows: starting injection voltage at −80 mV, recording time of 500 ms, with increments of 10 mV for eight sweeps down to 0 mV. All data are recorded using the MultiClamp 700B amplifier, with a sampling frequency of 5 kHz. The filtering frequency is set at 1 kHz to filter interference currents. P/N leak subtraction, leak current removal, and current analysis are performed using Clampfit 11.
The solution is listed below: (1) Cesium methanesulfonate electrode internal solution preparation: Cesium methanesulfonate: 125 mM CsCl: 5 mM Hepes: 10 mM EGTA: 0.2 mM MgCl2: 1 mM Mg-ATP: 4 mM Na-GTP: 0.3 mM Phosphocreatine disodium salt hydrate: 10 mM QX314: 5 mM pH: 7.30 Osmolality: 280 mOsm/kg. (2) Potassium gluconate electrode internal solution preparation: K-gluconate: 120 mM KCl: 6 mM Hepes: 10 mM EGTA: 0.1 mM Mg-ATP: 4 mM Na-GTP: 0.3 mM pH: 7.30 Osmolality: 280 mOsm/kg. (3) Cesium chloride electrode internal solution preparation: CsCl: 140 mM Hepes: 10 mM EGTA: 0.2 mM Mg-ATP: 4 mM Na-GTP: 0.3 mM QX314: 5 mM MgCl2: 1 mM pH: 7.30 Osmolality: 280 mOsm/kg.
Immunofluorescence
The cells were seeded onto glass slides coated with Matrigel and then treated with 4% paraformaldehyde (PFA) for 20 min at room temperature. Subsequently, they were permeabilized with 0.2% Triton-100 for 1 h and blocked with 1% bovine serum albumin (BSA) for 1 h at room temperature. The cocultured tissues were extracted from the solution at the specified time and preserved in a 4% PFA solution for 30 min at room temperature. Subsequently, they were treated with 0.8% Triton-100 and 3% BSA for 1 h at room temperature. Then, the cells or tissues were subjected to overnight incubation at 4°C in the primary antibody (Table 1). The second antibody was incubated for 1 h at room temperature, and protected from light (Table 2). The cell nuclei were stained with DAPI at room temperature for a duration of 15 min. To mitigate the influence of spontaneous background fluorescence on the results of cells and tissues with inherent biological properties, the sample was thoroughly inspected before incubating the primary antibody to ensure that the antigen did not display any signal. In all immunofluorescence staining experiments, a negative control was employed using a universal antibody diluent (NCM Biotech, WB100D) instead of the primary antibody to incubate the samples. Subsequently, the same immunofluorescence steps were carried out for the negative controls in the exact manner as in the experimental group. Images are photographed by an inverted fluorescence microscope (OLYMPUS, CKX53) or laser confocal microscope (Leica TCS-SP8; Tables 1 and 2).
Primary antibody list.

Secondary antibody list.

RNA extraction and RT-PCR
The total RNAs of hESCs-H9-ChR2-eYFP cells, pre-placodal cells (day 7), eONPs (day 19), and eONs (day 45) were extracted using the RNA-quick Purification Kit (RN001, ES Science) following the provided instructions. Subsequently, the extracted RNAs were converted into cDNA using the manufacturer’s protocols (11141ES60, Yeasen). The SYBR Green Master Mix (11201ES08, Yeasen) was employed in the RT-PCR analysis, utilizing the primers specified in Table 3. The Ct values were documented using the 7500 Real-time PCR system from Applied Biosystem, USA. The relative expression levels of the analyzed genes were determined using the formula: 2−ΔΔCt.
Primers for quantitative PCR.

Statistical analyses
The statistical analysis was conducted using an unpaired Student’s t-test and one-way ANOVA, as indicated. Values are commonly represented as mean ± standard error.
Results
Induction of hESCs to an auditory fate
Cells with an auditory fate were derived from hESCs through the manipulation of the cellular environment, with the application of a sequence of spatiotemporal growth agents, as depicted diagrammatically in Figure 1(a). The aim of the induction technique is to simulate key developmental stages of human spiral ganglion neurons (SGNs) in hESCs. Similar to the preceding review, this induction is divided comparably, including the initiation and growth of pre-placode (PP), the initiation and proliferation of embryonic stem cell-derived otic neural progenitors (eONPs), and the maturation of embryonic stem cell-derived otic neurons (eONs).

Generating otic neural progenitors (ONPs) and otic neurons (ONs) from hESCs. (a) Schematic representation of the protocol for generating otic neural progenitors (ONPs) and otic neurons (ONs) from hESCs. The timeline at the top displays the different time points, with the blue section indicating various stages in the induction process from hESCs to eONs, including PP induction, ONP induction and ON differentiation, and the green section representing the application of small molecule inhibitors LDN-193189 2HCl and IWP-2. Cytokines used during ONP induction and ON differentiation are shown in gray, and basal medium is shown in orange at the bottom with BGM at day 0 of cell induction, and DFNB as basal medium starting at day 13, respectively. The induced eONPs can be purified after 19 days, and the purified eONPs can be propagated during a period of 10 passages (more than 2 months). After purification was completed, DFNB was used as the basal medium in the differentiation stage of ONs, with the cytokines added as described in the flow chart. (b) Phase-contrast photomicrograph of hESCs-derived cells in different stages. It individually represents in the following stages as cells before induction (a), in PP stage (b), ONP stage (c), ON stage in lower resolution (d), and ON stage in higher resolution to show SGN-like neurons(e). (Scale bar: a–d, 100 μm; e, 25 μm. (c): Real-time quantitative PCR analysis of gene expression during induction. The graphs compare the expression levels of pluripotency markers Nanog (a) and OCT4 (b) between hESCs and PP cells. The expression of otic lineage markers PAX2 (c), EYA1 (d), SIX1 (e), GATA3 (f), and FOXG1 (g) is compared between hESCs, PP cells, and eONPs. Statistical significance is indicated by asterisks (a, n = 4; b, n = 6; c, n = 5; d-f, n = 4; g, n = 8; ****p < 0.0001; ***p < 0.001; **p < 0.01; *p < 0.05; a, b, t-test; c–g, one-way ANOVA). BDNF: Brain-Derived Neurotrophic Factor; BGM: Basic growth medium; DFNB: DMEM/F12/N2/B27 medium; DM: differentiation medium; GDNF: glial cell line-derived neurotrophic factor; IWP-2: Wnt pathway inhibitor; LDN-193189 2HCl: BMP Signaling Inhibitors; NT3: Human Neurotrophin-3; ON: otic neuron; ONP: otic neural progenitor; PP: pre-Placode; rhFGF3: Recombinant human fibroblast growth factor3; rhFGF10: Recombinant human fibroblast growth factor10; β-NGF: Nerve Growth Factor-β;
Based on dual SMAD inhibition, the small molecule inhibitors LDN193189 2HCl and IWP-2 as well as SB431542 initiated the differentiation of hESCs. 27 During this period, dense clones of hESCs underwent a transition to a more dispersed state, and the shape of the cells became flattened. Quantitative polymerase chain reaction (qPCR) analysis revealed a significant decrease in the expression of pluripotent genes Nanog and OCT4 after the PP stage (Figure 1(c)-a, (c)-b; p < 0.0001, t-test). Additionally, the expression levels of pre-placodal markers PAX2, EYA1, and SIX1 showed a significant increase compared to hESCs (p < 0.0001, p < 0.0001, p < 0.001, one-way ANOVA; Figure 1(c) c-e). Immunofluorescence staining revealed that the cells exhibited positive expression for SIX1 and PAX6, whereas the ESC marker OCT4 was no longer detected in the presumed PP stage (Supplemental Figure 2A). Taken together, these data demonstrated that by modulating specific pathways, specifically the dual SMAD pathway, hESCs were stimulated to differentiate into pre-placodal cells.
Prior studies have revealed that the FGF family plays a vital role in the development of the inner ear, and hESCs have a tendency toward becoming inner ear cells through the control of FGF.23,39 Therefore, we hypothesized that by continuously introducing FGF3 and FGF10, cells at the PP stage can be progressively induced into otic neural progenitors, which will ultimately develop into SGN-like cells. To test this hypothesis, cells were cultured in an induction medium with the addition of FGF3 and FGF10 after the pre-placodal induction stage. 23 Following a 12-day treatment, the cells displayed a more elongated and spindle-shaped morphology, characterized by reduced cell size and a more intense cytoplasmic coloration. Certain cells showed a propensity for elongating their process (Figure 1(b)-c). On the 19th day, the levels of SIX1, GATA3, and FOXG1 expression were found to increase significantly according to qPCR analysis (Figure 1(c) e-g). The immunofluorescence staining technique was used to detect the proportion of cells expressing SIX1 and PAX6, as shown in Figure 2(a). The successful induction of SIX1 expression was observed in 16.81% ± 2.57% of cells (Figure 2(c)). The qPCR and immunostaining data indicated that the induced cells may acquire the fate of auditory progenitors.

Characterization of otic neural progenitors (ONPs) derived from hESCs before and after purification. (a) Immunofluorescence images demonstrating the expression of PAX6 and SIX1 in eONPs before and after purification. In the ONP stage, PAX6+ cells occupy almost 80% of cells before purification, and PAX6+ cells reached 92% ± 2% after purification. About 20% of the cells expressed SIX1 before purification, and SIX1+ cells reached 80% ± 3% after purification. Scale bars: 100 μm. (b) and (c) Quantification of PAX6 (b) and SIX1 (c) expression levels in eONPs before and after purification using collagenase IV, as determined by immunocytochemistry staining (n = 4; ****p < 0.0001; **p < 0.01; t-test). (d) Immunofluorescence staining of eONPs for various neural progenitor markers: FOXG1 (a), PAX2 (b), Brn3A (c), NESTIN (d), PAX8 (e), and TUJ1 (f). Scale bars: 100 μm. (e) Diagram of quantification illustrating the expression proportions of ONP markers (FOXG1, PAX2, Brn3A, TUJ1, NESTIN, and PAX8) as determined by immunostaining of purified eONPs at day 19 of differentiation. (n = 4).
The purity and proportion of eONPs play a crucial role in enhancing the impact of eONs on differentiation and coculture. Following a 19-day induction protocol of hESCs, the resultant heterogeneous cell population—comprising presumptive eONPs or SIX1+ cells and other cell types—was subjected to selective enzymatic treatment with collagenase IV at 37°C. This treatment exploits the differential enzymatic sensitivity among cell types, thereby facilitating the selective dissociation of non-target cells. The non-ONP cells, which are more vulnerable to collagenase IV, were preferentially detached and eliminated, thereby enriching the population with high-purity eONPs or SIX1+ cells.
The digestion process can be then observed using a microscope (Supplemental Figure 3). Following the FGF induction, the SIX1+ cells formed clusters and proliferated collectively in culture when observed under the microscope. The SIX1+ cells or eONPs and other cell types were separated at different times and in different sequences. Upon the application of collagenase IV to the adherent cells, the entire purification process spanned approximately 1 h, with observations made at 5-min intervals. Microscopic examination revealed that the non-target cells progressively contracted into clusters under the influence of collagenase IV and eventually detached from the culture flask bottom, as depicted in Supplemental Figure 3a–d. After the removal of these impurities, the eONPs remained adherent, as shown in Supplemental Figure 3e. The eONPs were subsequently dissociated into single cells using Accutase (Supplemental Figure 3f) and were then ready for further experimental applications such as passaging, differentiation, and immunofluorescence.
To evaluate the effectiveness of differential digestion in enriching the population of eONPs, the expression of ONP markers, SIX1 and PAX6, was examined. After purification with differential digestion, the proportion of SIX1+ and PAX6+ cells increased significantly. Notably, the population of PAX6+ cells, which comprised approximately 80% of the pre-purification population, increased to 92% ± 2% after purification. Similarly, the percentage of SIX1+ cells exhibited a substantial increase, rising from 20% before purification to 80% ± 3% post-purification (Figure 2(b) and (c)). These findings suggest that differential digestion serves as an efficient method for enriching the eONP population.
PAX2 and PAX8 were expressed in 96.94% ± 1.27% and 73.98% ± 1.97% cells separately after purification. Nestin and FOXG1 were also expressed in most cells (80.16% ± 3.57%, 64.79% ± 11.64%). The expression of pan-neural marker TUJ1 was 89.28% ± 2.22% of cells and that of sensory neural marker Brn3A was found as 92.46% ± 4.73% of cells (Figure 2(d) and (e)). Taken together, these data suggested that eONPs with high purity were obtained successfully from induced cells following ONP induction and proliferation. Our qPCR and immunostaining data showed that eONPs were obtained successfully from PP cells, in addition, ONP fate was adopted based on the ICC staining result of ONP markers expression and related neuronal markers.
eONs obtained from eONPs
With the aim of obtaining eONs, we cultured the eONPs in the differentiation medium for a period of 1–2 weeks of differentiation from the ONP stage to the expected stage of ONs. Typically, at this stage, the maturation of eONs could be observed based on the increased cell polarity, more rounded and plump cell bodies. The differentiation process was then terminated and thereafter proceeded with identification studies or coculture experiments.
The immunostaining results demonstrated positive expression of the neuron markers neurofilament (NF200) and β-III tubulin, as well as the mature neuron marker MAP2, in the differentiated cells (Supplemental Figure 4). The expression of more specific markers of human SGNs, including peripherin (PERI),40,41 TrkB, and TrkC receptors, was also identified in these cells (Supplemental Figure 4, Figure 3(a)-b, (a)-c). The expression of SYN and VGLUT1 provided evidence for the differentiation of glutamatergic neuronal fate (Figure 3(a)-d, (a)-e). The expression levels of Ki67 decreased by 20.05% ± 4.995% compared to eONPs, suggesting a departure from the cell cycle. Nevertheless, a subset of the cells exhibited Ki67 positivity, suggesting that they may still be in the progenitor stage (Supplemental Figure 5A, B). The real-time qPCR analysis demonstrated a significant increase in the expressions of N-cam, TUBB3, TRKB, TRKC, and VGLUT compared to the ONP stage. These findings confirm the similar results observed in immunofluorescence staining at the mRNA level (Figure 3(b)).

Immunofluorescence and Electrophysiological Evaluations of eONs Differentiated from eONPs. (a) Immunocytochemistry staining of SGN-like glutamatergic neurons derived from eONPs. The images displayed the expression of neuronal markers TUJ1 (a), tyrosine kinase receptors 2/3 (TrkB and TrkC; b, c), Synapsin 1 (a synaptic vesicle protein; d), and VGLUT1 (vesicular glutamate transporter; e). Scale bars: 25 μm. (b) Quantitative analysis of gene expression in ONP and ON stage using real-time PCR. The graph depicts the relative expression levels of neural markers N-cam (a), TUBB3 (b), TRKB (c), TRKC (d), and VGLUT (e). Statistical significance is indicated by asterisks (a-c, e, n = 4; d, n = 8; ****p < 0.0001, t-test). (c) Electrophysiological properties of eONs assessed by patch-clamp recordings. The graphs illustrate various parameters including: representative traces of action potentials (a, b), peak potential (c; n = 5), resting membrane potential (d; n = 5), threshold potential (e; n = 6), half length of action potentials (f; n = 5), time constant (g; n = 5), and input resistance (h; n = 5).
To further assess the long-term stability and differentiation potential of eONPs, we conducted extended proliferation assays. eONPs were maintained in DFNB medium supplemented with FGF3 and FGF10 for over 10 passages, spanning a period exceeding 60 days. Following this extended expansion phase, the proliferative eONPs were subjected to the differentiation protocol for 1–2 weeks. Immunofluorescence analysis of the differentiated progeny revealed robust expression of neuronal markers TUJ1, TRKB, and TRKC, alongside the glutamatergic neuronal marker VGLUT (Supplemental Figure 6). The presence of these characteristic markers confirms the successful differentiation of expanded eONPs into cells exhibiting a SGN-like identity. These findings demonstrated that eONPs retained their capacity to differentiate into eONs even after prolonged in vitro expansion, highlighting their potential for generating a substantial pool of SGN-like cells for downstream applications.
Collectively, these data led us to conclude that the cells had successfully adopted the cell fate of auditory neurons.
We employed the patch-clamp technique to measure the action potentials of the generated differentiated eONs in order to determine their level of maturity. Figure 3(c)-a and (c)-b shows that action potentials were generated by eONs, similar to those observed in vivo in SGNs. 42 We performed calculations for various electrophysiological parameters, including peak potential (Figure 3(c)-c), resting membrane potential (Figure 3(c)-d), threshold potential (Figure 3(c)-e), half length (Figure 3(c)-f), time constant (Figure 3(c)-g), and input resistance (Figure 3(c)-h). The average values of these parameters were found to be similar to those observed in live organisms. This indicates that stem cell-derived ONs possess similar electrophysiological characteristics to SGNs. 27
Coculture of eONs and cochlear nuclear neurons
To visually monitor and evaluate the morphological and differentiation characteristics of the cocultured eONs, we developed a genetically modified hESC line, hESCs-H9-EGFP, which was created through electroporation-mediated insertion of the EGFP gene (Figures 4(a) and Supplemental Figure 7). To further enhance our ability to study the functional characteristics of hESC-derived auditory neurons, we engineered additional hESC lines with optogenetic and fluorescent capabilities. The plasmid expression vector containing channelrhodopsin-2 (ChR2)-eYFP was introduced into hESCs-H9 lines using lentivirus infection (Supplemental Figure 8), resulting in the creation of a cell line called hESCs-H9-ChR2-eYFP, as displayed in Figure 4(b). This innovative approach allows for the precise optical control of neuronal activity, thereby facilitating advanced studies on neuronal connectivity and function within our coculture system. Then, ONPs induced from hESCs cells expressed ChR2-eYFP or EGFP and differentiated into eONs in DM medium after 7–14 days.

Establishing a coculture system of eONs-ChR2-eYFP and CNNs. (a) Microscopic visualization of the hESC-H9-EGFP cell line. The cells exhibited robust green fluorescence due to the expression of the EGFP reporter gene. Scale bars: 100 μm. (b) Microscopic visualization of the hESC-H9-ChR2-eYFP cell line. The image presented both phase contrast and fluorescence channels. The cells exhibited fluorescence under the inverted fluorescence microscope (OLYMPUS, CKX53) due to the expression of the eYFP reporter gene, suggesting the presence of the ChR2(H134R) optogenetic tool within the engineered cells. Scale bars: 100 μm. (c) Schematic illustration of the process for obtaining anteroventral cochlear nucleus (AVCN) cells from the cochlear nucleus and establishing a coculture system with eONs. (d) Immunofluorescence image demonstrating the expression of P2X2 receptors in isolated CNNs. Scale bars: 100 μm. (e) Phase-contrast and fluorescence microscopy images of cocultured CNNs and eONs-EGFP after 10 days. eONs-EGFP were cocultured with EGFP-negative rat AVCN cells (indicated by yellow arrows). The images reveal the formation of neurite connections between the two cell types. Scale bars: 50 μm.
To determine whether functional connections could be established between the induced SGN-like eONs and cochlear nucleus neurons (CNNs), the induced eONs and CNNs were cocultured together. CNNs were extracted bilaterally from the anterior ventral region of the medulla oblongata in 12-day-old rats. Following the coculture of CNNs and induced eONs-ChR2-eYFP or eONs-EGFP for a period of 7–10 days, the functional connectivity between them was assessed by stimulating blue light at a wavelength of 473 nm (Figure 4(c)). The Cultured AVCN cells displayed spherical cell bodies with short, tufted dendrites and expressed the P2X2 receptor, a marker for CNNs (Figure 4(d)). 43 After 10-day coculturing of eONs-EGFP and CNNs, eONs projected nerve fibers to CNNs and resulting in the formation of a reticular neural structure connecting them (Figure 4(e)).
The immunofluorescence results showed that cocultured eONs-EGFP and CNNs could express Synapsin 1 (Figure 5(a)-a), a marker of synaptic proteins. EGFP+ eONs formed a structural connection with EGFP− CNNs, and the nerve fibers of eONs were shown to overlap with the neurites of CNNs (Figure 5(a)). The positive immunostaining of VGLUT indicated that the transport mode of this synapse was glutamine-based (Figure 5(a)). The expression of TrkB and TrkC was also detected in EGFP+ eONs, which were shown to connect the CNNs based on the immunostaining results shown in Figure 5(a), (a)-d.
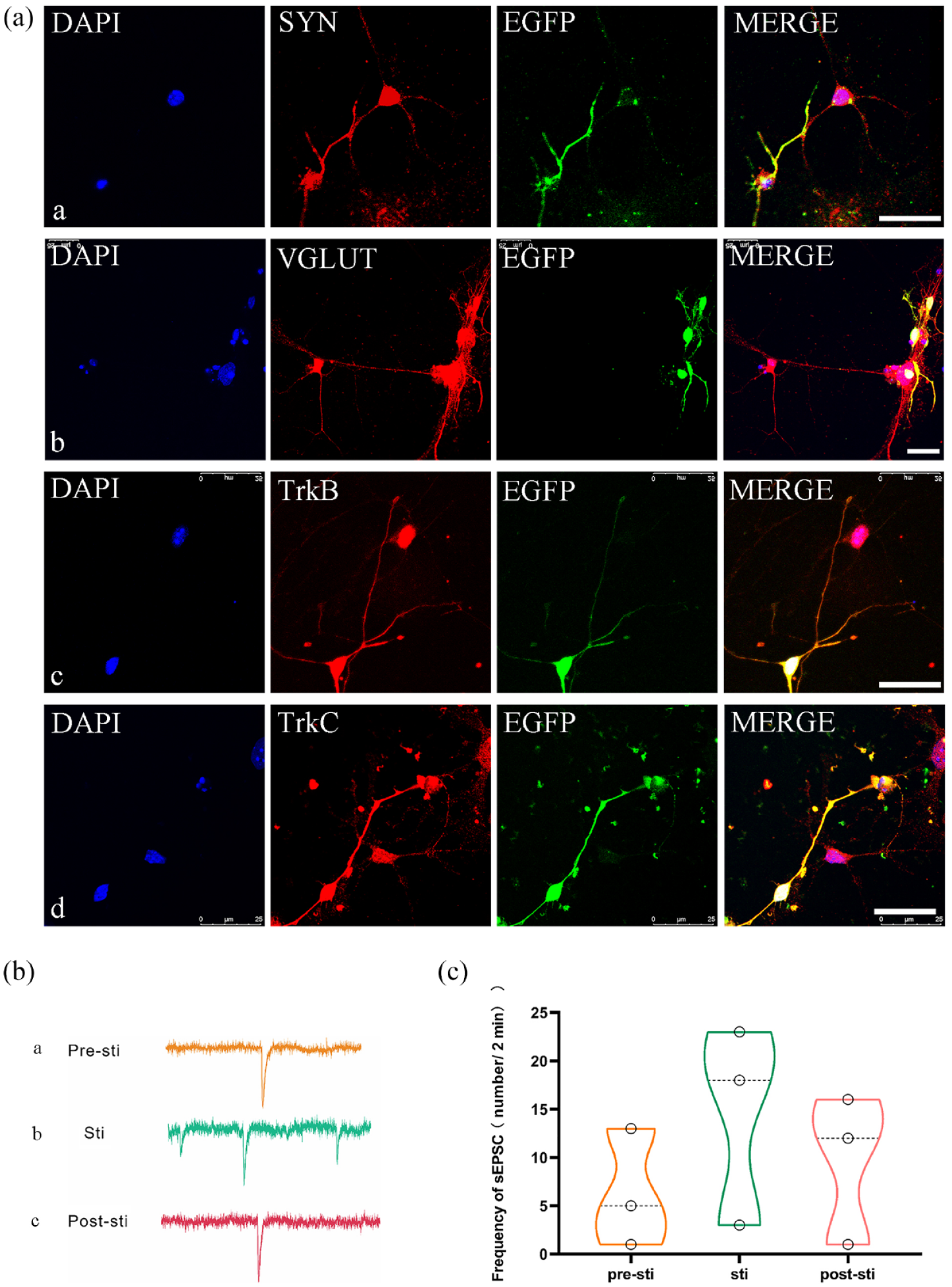
Functional synapse formation and optogenetic stimulation in eON-CNN cocultures. (a) Immunofluorescence analysis of cocultured cochlear nucleus neurons (CNNs) and induced eONs-EGFP after 10 days. The images demonstrated the expression of various markers in both cell types: SYN (synapsin-1; a), VGLUT (b), TrkB (c), and TrkC (d). Furthermore, the images highlighted the formation of connections between the neural fibers of eONs-EGFP and EGFP-W/O (without) CNNs. Scale bar: 25 μm. (b) Electrophysiological recordings of CNNs cocultured with eONs-ChR2-eYFP. The pictures illustrated the electrical activity of CNNs (a) before, (b) during continuous light stimulation of eONs-ChR2-eYFP, and (c) after light stimulation. (c) Quantification of spontaneous excitatory postsynaptic currents (sEPSCs) in CNNs. Continuous exposure to light in the eONs-ChR2-eYFP resulted in a significant increase in the frequency of sEPSCs in the downstream CNNs (n = 3, p < 0.01, one-way ANOVA). The frequency of sEPSCs in CNNs decreased after cessation of light stimulation (n = 3, p < 0.05, one-way ANOVA).
To confirm the functionality of synapses between eONs and CNNs, CNNs, and eONs-ChR2-eYFP were cocultured and the patch-clamp technique was employed to quantify the alterations in EPSC frequency in CNNs coculture. Figure 5(b) and (c) demonstrated that continuous light stimulation of upstream eONs increased the frequency of EPSCs of downstream CNNs (p < 0.01). Meanwhile, once the light stimulation was removed, the frequency of sEPSCs in downstream CNNs decreased (p < 0.05). These findings indicated that the light-evoked upstream eONs could deliver neurotransmitters to downstream CNNs, and functional connections were confirmed by optogenetic stimulation.
Coculture of eON spheres and CN-BS
To assess the potential integration of eONs with CNNs for future in vivo applications, we obtained cochlear nucleus tissue from brain slices and cocultured it with the eONs at the organ level. The eON-EGFP spheres and cochlear nucleus taken from brain slices were cocultured together on the semi-permeable membrane (Figure 6(a)). The process was observed to spread out gradually from eON-EGFP spheres and nerve fibers were projected to CN-BS over time (Figure 6(b) c–e). Both TUJ1 and VGLUT+ were found to be expressed in cocultured eONs and tissues (Figure 6(c)). Neurites that express Synapsin 1 and VGLUT were shown to extend from EGFP+ exogenous eONs to polysynaptic cells of the natural cochlear nuclei in the brain slice (Figure 7(a)). These results indicated that eONs could establish structural connections with the cochlear nucleus at the organotypic slice.

Investigation of connection by the coculture of CN-BS and eON-EGFP spheres. (a) Schematic representation of the coculture system, where CN-BS and induced eON-EGFP spheres were cultured together to study their interactions and potential synapse formation. (b) Phase contrast and fluorescence microscopy images of CN-BS and eON-EGFP spheres after a specific coculture period. These images provide an overview of the overall structure and distribution of the cells within the coculture system. Scale bar: a–c, 500 μm; d, 200 μm; and e, 100 μm. (c) Immunofluorescence staining of CN-BS and eON-EGFP spheres cocultured for 7–10 days. The images revealed the expression of neuronal marker TUJ1 and synaptic marker VGLUT in both CN-BS and eON-EGFP spheres. The green fibers contact with ungreen fibers, and in the same time VGLUT and TUJ1 exrpessed in both cell fibers. It suggest the formation of synaptic connection. Green arrows indicate eONs-EGFP, while red and purple arrows point to TUJ1-positive and VGLUT-positive neurons of CN-BS, respectively. The images showed the establishment of structural connections between eONs and neurons of CN-BS at different magnifications. Scale bar: a, 200 μm and b, 25 μm.

Synaptic protein expression and optogenetic activation in coculture of eON spheres and CN-BS. (a): Immunofluorescence staining of cocultured CN-BS and eON-EGFP spheres. The images displayed the expression of synaptic proteins Synapsin1 and VGLUT, indicating the formation of synapses between the two cell types. DAPI staining was used to visualize cell nuclei. Scale bar: 25 μm. (b) Optogenetic activation of neurons of CN-BS cocultured with eONs-ChR2-eYFP spheres. (a–d): Blue light stimulation of eONs-ChR2-eYFP spheres led to a significant increase in the levels of INa+/IK+ (ratio of sodium to potassium current) in neurons of CN-BS (n = 5, INa+ p < 0.05, IK+ p < 0.01, t-test), demonstrating functional synaptic transmission and activation of neurons of CN-BS upon light-induced depolarization of eONs. (e, f): Representative traces of action potentials recorded from neurons of CN-BS before and after blue light stimulation (e). The increased counts of action potentials in neurons of CN-BS after light stimulation further confirmed the functional connectivity between eONs and neurons of CN-BS (f; n = 5, p < 0.01, t-test).
To investigate whether eONs could form potentially functional synaptic connections with cochlear nuclei in a simulated intracerebral environment, we conducted electrophysiological recordings of CNNs in brain slices following 10 days of coculture with eONs-ChR2-eYFP. Following light stimulation, there was a significant increase in both INa+/IK+ of downstream cochlear nucleus neurons (p < 0.05; Figure 7(b) a–d). Action potentials were recorded to further confirm the functionalities of these cellular synaptic connections. No action potentials could be observed from downstream neurons without light stimulation. After light stimulation of upstream eONs-ChR2-eYFP, we found increased numbers of action potentials released in downstream neurons (Figure 7(b) e, f). These electrophysiological results suggested that eONs could form stable and functional synaptic connections with the cochlear nucleus within the simulated intracerebral environment.
Discussion
Fundamentally, stem cell therapy holds promise for treating neurosensory hearing loss.23,44,45 It is still a challenge to isolate and derive enough and appropriate primary stem cells from patients for cell therapy, whereas pluripotent stem cells including hESCs and hiPSCs can provide an adequate source of cells.23,42,46 hESCs were induced into SGN-like cells using a different technique based on the principles of developmental biology.
In addition to the differentiation of stem cells from non-ESCs,17,21 we induced hESCs into SGN-like cells. hESCs can be induced into otic neural progenitors under the action of FGF3 and FGF10, which are important signals involved in early inner ear development. Treatment with FGF3 and FGF10 produced global changes in transcription that were consistent with the induction of otic progenitor cells. 23
Research has shown that the co-administration of WNT agonist and TGFβ/SMAD inhibitors leads to a notable enhancement in the development of existential neurospheres, as well as the potential to grow auditory neural precursors 47 and induce ONPs. 27 SGN-like cells were gradually induced and differentiated without the use of FGF3 and FGF10 by mimicking the development and differentiation of SGNs in vivo. 27
In our study, the differentiation stage is similar to that of SGN-like cells induced by ESCs, which is divided as: non-neural ectoderm, otic placode and otocyst, neuroblast, and SGN-like neurons. 48 In PP stage, hESCs were induced into preplacode cells, in which cells exhibited specific morphology, and the expression of hESCs pluripotent marker decreased, while the expression of preplacode marker increased. Then, the cells were induced into eONPs committing to auditory fate under the action of FGF3 and FGF10, after which eONPs can be maintained and proliferated for more than 10 passage (60–90 days, depending on batch). With or without the ONP proliferation stage, eONPs can differentiate into SGN-like neurons after 1–2 weeks, which exhibited the expression of SGN markers and some electrophysiological activity. To sum up, our method is based on the principle of early auditory development, and the phenotype and electrophysiological activity of differentiated SGN-like cells are similar to those of SGNs in vivo.
Isolation of pure cell population is crucial for stem cell biology research. 49 Similarly, to purify NSCs is the priority for NSC-based cell therapy. The differentiation of NSCs may be impacted by contamination or mixing with other cells, which could result in side effects and impede the translational process. 50
Otic progenitors had been induced from hPSCs and purified by Fluorescence-activated cell sorting (FACS), according to Matsuoka et al. 27 Nevertheless, during fluorescence-activated cell sorting, cells are subjected to a variety of stresses, including high pressure and decompression, acceleration, high speed, forces, laser illumination, electrical charge, and rapid temperature change. These forces can change cellular structure and function, trigger stress pathways, and impact cell metabolism.51,52 Any of these could lead to modifications in the health, vitality, and gene expression of cells.
As we previously reported, NSCs from various sources were used for nerve regeneration, including the auditory, 23 enteric,53,54 and sciatic nerve. 55 These NSCs are all extremely pure, FACS-free, and able to restore the relevant function in organ or animal models. Here, we present an easy-to-implement protocol that is based on the ideas of early auditory development and does not require more harmful steps, like FACS. The induced cells in the ONP stage are a mixed population, with SIX1+ cells making up a portion of the cell population on day 19. Collagenase IV was used to dissociate different cell populations at different times and sequences in order to identify and isolate SIX1+ cells and other cell types in the ONP stage.
In contrast to other cell types that exhibit in a dispersed and individual way, SIX1+ cell population can stay unaltered in a petri dish for a longer period of time before being detached by Collagenase IV. High-purity SIX1+ cells or supposed eONPs were thus produced, and a significant fraction of differentiated eONs in cell culture are also derived from SIX1+ progenitor cells.
To sum up, the straightforward induction protocol for eONPs is based on the principle of early auditory development. It eliminates the need for flow cell sorting and other potentially harmful steps, and the resultant eONPs and eONs have a high degree of purity.
ONPs derived from PSCs were demonstrated to exhibit integration, differentiation, and cell survival after transplantation.22,23,28–31 On the other hand, the functional integration of exogenous NPs into auditory circuit has been scarcely elucidated. We have reported that hESCs can be used to induce otic neural progenitors, which can then be readily isolated and differentiated into SGN-like cells exhibiting electrophysiological activity. The ABR of hearing of auditory neuropathy model was restored after transplantation. 23 Nevertheless, due to the limitations of Ouabain modeling, the functional recovery following ONP transplantation does not completely rule out the possible influence of a few residual SGN cells that might persist in the animal model. More research is required to determine whether these cells could integrate functionally into the auditory circuit.
Previous studies have shown that SGN damage usually leads to the dysfunction of transmitting auditory signals to the brain stem. In rodents, principal cells of the ventral cochlear nucleus receive auditory signals from SGN.11,56 Therefore, the ability to communicate with the CNNs is crucial in order to ascertain whether exogenous auditory neurons can be employed in the treatment of SNHL.
In vitro coculture system can mimic the in vivo environment to a large extent. Application of coculture is possible to clarify the interaction mechanism between neural cells and replicate neural circuits in vitro. The in vitro coculture model offers a more practical and accurate way to illustrate the mechanism of interaction between cells, tissues, or organs than does the animal model. 57 To investigate whether synapses and electrophysiological activity could be exhibited between cochlear nucleus neurons and mESC-derived SGN-like cells, a cellular coculture system was utilized 32 .
Cao et al. 56 found that properties of synapses between spiral ganglion cells and target cells exhibited systematic differences. Accordingly, it was indicated that coculture system of more principal cell types from the ventral CN (VCN) with exogenous cells needs to be established for further research. From the ventral cochlear nucleus, we isolated a population of representative multipolar cells that expressed the P2X2 receptor. The outcomes of the experiment demonstrated that these CNNs could be effectively connected to the induced auditory neurons.
If newly formed synapses are functional and able to transmit electrical signals, this can be ascertained by electrophysiological studies. 58 With traditional electrophysiological techniques, it is challenging and time-consuming to verify that two cultured cells are linked and have an upstream and downstream relationship in the circuit. This requires dual patch-clamp recordings of the cells. Electrodes could be used to stimulate one or more cells simultaneously, and electrical field stimulation could make it more difficult to interpret the results.
In some cases, selective stimulation of a specific subset of cells is urgently necessary. To address this need, optogenetic techniques pre-label cells with light-sensitive ion channels, providing a means of confirming the functional relationship between different parts of the neural circuitry.34–38 Neurons became excited and generated an electrical signal when the light-sensitive ion channel ChR2 was exposed to light stimuli.34,35 Nevertheless, the potential circuitry between exogenous neurons and CN or CNNs has not been assessed using this method.
The auditory circuit’s hair cells convert mechanical signals into electrical signals. Sent to the cochlear nucleus, SGNs generated an electrophysiological response upon receiving it.59,60 Cochlear implants and hair cells can still produce electrical signals when SGN is damaged, but they cannot reach CN. Stem cells represent a promising therapeutic approach for repairing the damaged sensory circuitry. In this study, ONs- ChR2-eYFP were generated from hESCs and, when exposed to blue light, generated an afferent upstream signal resembling that of hair cells.
We showed that functional neural connections were formed between CN and SGN-like neurons at the cell and organ level in coculture using optogenetic technology. We showed that when eONs-ChR2-eYFP were cocultured with rodent CNNs and an organotypic CN-BS, light-evoked electrophysiological responses were generated. This work offers important evidence that eONs are capable of building auditory circuits with cochlear nuclei and show promise for SNHL treatment.
In neurological functional studies, brain slices are frequently employed. 61 Compared to cell culture, organotypic slices can maintain supporting cells, glial cells, immune cells, etc. thus having a more complex environment and are closer to the vivo environment. 62 Therefore, the ability to establish neural connections with organotypic brain slices is an important aspect of assessing exogenous cells for cell therapy. 63 It was reported that mouse ESCs could be differentiated toward neurons when cocultured with brain slices. 64
In this study, hESCs-derived SGN-like neurons in coculture with organotypic slice projected neuronal process into the slice. Functional studies have shown that the differentiated auditory neurons, modified by light-sensitive channels, could generate light-evoked excitation upon excitation light irradiation and transmit this signal to central nervous nucleus. Additionally, sodium and potassium currents contribute to maintaining both the excitatory transmission of the synaptic connections and the stability of membrane potentials. These mechanisms ensure a balance between excitability and stability for proper neuronal communication. Light-evoked action potentials were detected in the CN-BS, which marked the successful completion of the step-by-step conversion of light-electric-chemical-electric signals. This research validates that neurons derived from hESCs are capable of forming connections with animal organotypic slices, providing further experimental support for their subsequent studies in vivo.
The next goal is to explore whether eONs can integrate with cochlear implants in clinical applications. The ultimate goal is to integrate otic neurons or otic neural progenitors into the auditory circuit structurally and functionally in clinical application. This process could potentially restore or enhance hearing capabilities in patients with auditory impairments.
Conclusion
eONs have similar biochemical and electrophysiological characteristics to SGNs and can establish structural and functional connections with CNNs and organotypic CN-BS, suggesting that they can be a potential candidate source of cell therapy for the treatment of SNHL.
Supplemental Material
sj-docx-1-tej-10.1177_20417314241265198 – Supplemental material for Optogenetically modified human embryonic stem cell-derived otic neurons establish functional synaptic connection with cochlear nuclei
Supplemental material, sj-docx-1-tej-10.1177_20417314241265198 for Optogenetically modified human embryonic stem cell-derived otic neurons establish functional synaptic connection with cochlear nuclei by Yanni Chen, Wenbo Mu, Yongkang Wu, Jiake Xu, Xiaofang Li, Hui Hu, Siqi Wang, Dali Wang, Bin Hui, Lang Wang, Yi Dong and Wei Chen in Journal of Tissue Engineering
Footnotes
Acknowledgements
The authors thank members of the technical platform at the Collaborative Research Center, Shanghai University of Medicine and Health Sciences for helpful advice and comments.
Abbreviations
ACSF, Artificial Cerebrospinal Fluid; AP, action potential; AVCN, anteroventral cochlear nucleus; ChR2, Channelrhodopsin-2; CN, cochlear nucleus; CNN, cochlear nucleus neuron; CN-BS, cochlear nucleus of brain slices; eONPs, embryonic stem cell-derived otic neural progenitors; eONs, embryonic stem cell-derived otic neurons; hESCs, human embryonic stem cells; ONPs, otic neural progenitors; ONs, otic neurons; PNS, peripheral nervous system; PP, pre-placodal; sEPSC, spontaneous excitatory postsynaptic currents; SNHL, sensorineural hearing loss; SGN, Spiral ganglion neuron; TRKB, Tropomyosin receptor kinase B; TRKC, Tropomyosin receptor kinase C; TUBB3, Tubulin Beta 3 Class III.
Author contributions
Y.C., W.M., and Y.W.: experimental design, collection and assembly of data, data analysis and interpretation, and manuscript writing; J.X., X.L., H.H., and S.W.: collection and assembly of data, data analysis, and interpretation; D.W. and B. H.: provision of study material; L.W. and Y.D.: data analysis and interpretation, study supervision, manuscript writing, and final approval of manuscript; W.C.: conception and design, financial support, data analysis and interpretation, manuscript writing, study supervision, and final approval of manuscript.
Data availability statement
The data used to support the findings of this study are available from the corresponding author upon request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was financially supported by the National Natural Science Foundation of China (Grant No. 32170805), Shanghai Natural Science Foundation (Grant No. 23ZR1427300), Research Foundation for Talents in Shanghai University of Medicine & Health Sciences, and Research Foundation for Stem Cell and Organoid-Based Disease Prevention and Treatment in Shanghai University of Medicine & Health Sciences.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.