
Abstract
Recently, researchers have focused on the role of gut microbiota on human health and reported the existence of a bidirectional relationship between intestinal microbiota and the brain, referred to as microbiota-gut-brain axis (MGBA). In this context, the development of an organ-on-a-chip platform recapitulating the main players of the MGBA would help in the investigations of the biochemical mechanisms involved. In this work, we focused on the development of a new, hydrogel-based, 3D brain-like tissue model to be hosted in the brain compartment of the aforementioned platform. We previously cultured primary mouse microglial cells, cortical neurons and astrocytes independently, once embedded or covered by a millimeter layer of two selected collagen-based hydrogels. We evaluated cell metabolic activity up to 21 days, cell morphology, spatial distribution and synapse formation. Then, we exploited the best performing culturing condition and developed a more complex brain-like tissue model based on the co-culture of cortical neurons and glial cells in physiological conditions. The obtained results indicate that our 3D hydrogel-based brain tissue model is suitable to recapitulate in vitro the key biochemical parameters of brain tissue.
Introduction
Mammalian brain tissue is complex, and challenging to model in vitro. However, the investigations of several pathophysiologic mechanisms would greatly benefit from the availability of reliable cell-based systems featuring at least the main brain cell populations, neurons and glial cells. An example where such models could help move our knowledge forward is the microbiota-gut-brain axis (MGBA). In the last few years, the number of publications on the role of gut microbiota in human health has increased significantly and researchers have reported a bidirectional relationship between gut microbiota and the brain. It exploits several communication pathways, such as the release of microbial factors, intestinal hormones and cytokines from the immune system.1–4 In this context, the Authors aim at developing an optically accessible multi-organ-on-a-chip platform hosting advanced two- (2D), suspended, and three-dimensional (3D) in vitro models to recapitulate the main players of the MGBA, such as the microbiota, the gut, the immune system, the blood-brain barrier and the brain, in physiological and pathological conditions. 4 In the present work, we focused on the setting up of a millimeter-thick brain-like tissue model potentially suitable for dynamic culture in a recently developed optically accessible organ-on-a-chip (OOC) device. 5
In the last decade, OOC technology has seen considerable improvements for its potential to reduce animal studies for drug development and toxicity testing. 6 Its reliability and reproducibility offer substantial advantages for tissue and disease modeling,7,8 overcoming the limitations of 2D static cultures.
Despite their simplicity compared to the in vivo situation, 3D models have significantly contributed to the recapitulation of tissue properties, cell-cell contacts and cell-extracellular matrix (ECM) interactions.9–12 Organoids, miniaturized self-organized tissue cultures from stem cells, are among the most promising 3D models for brain representation.13,14 They have been employed to study brain development and organization, as well as neurological diseases. 15 However, they lack cell aging 16 and internal vascularization, inducing necrosis. 17 They are hard to grow, labor-intensive and time-consuming, all major disadvantages for investigations of disease pathways and drug targets. 18
As an alternative to organoids, hydrogels are excellent candidates to mimic the ECM of soft tissues and have emerged for the development of 3D brain-like tissue models. 19 Their stiffness is a key factor in the regulation of neuronal cell shape, viability, expression, migration and differentiation, both in 2D20,21 and 3D conditions. 22 Several studies have indicated that softer gels promote neurite outgrowth, 23 while glial cells prefer a stiffer microenvironment. 24 However, some studies reported that soft hydrogels support astrocyte differentiation and survival. 25
Microglia, the resident immune cells of the central nervous system (CNS), play a key role in the maintenance of CNS homeostasis and in the management of tissue response to injury. 26 They are involved in monitoring synapse extension 27 and remodeling during development, 28 they contribute to neuroprotection and regeneration by releasing cytokines, molecules and other neurotrophic factors. 29 Studies coupling microglia and hydrogels are not numerous. Up to now, they have focused on hydrogel effects on cell morphology, adhesion and motility, 26 and cytokine release. 30
Hydrogels have been used to culture neural cells in different 3D conditions. Ylä-Outinen et al. cultured human embryonic stem cell-derived neural cells up to four weeks under PuraMatrix ™ hydrogels. The cells were cultured on laminin-coated microplates for some days, then covered with a hydrogel layer, or encapsulated into the hydrogels after mixing with the polymer solution. 31 Xu et al. proposed a sandwich-based condition, in which the cells were grown at the interface of two hydrogel layers for 21 days. 32
For a long time, neurons have been cultured alone in biomaterials or devices and the role of glial cells has gone into the background. Now it has clearly emerged that the advanced modeling of brain-like tissues depends on the co-culture of different neural cell populations and the investigations of their interactions. 15 For instance, astrocytes play key roles in neural functions, such as axon growth and direction, synaptogenesis, formation of the blood-brain barrier, and inflammatory responses. 33 Their morphology, proliferation rate and marker expression are governed by 2D or 3D culture conditions, with 3D cultures providing conditions more similar to the in vivo situation and allowing early postnatal cells to transit to the differentiated stellate morphology. 33
Starting from the literature and our previous works with immortalized neuronal cells,34,35 we employed semi-interpenetrating polymer networks (semi-IPNs) prepared by promoting collagen (COLL) fibrillogenesis in the presence of hyaluronic acid (HA) or poly(ethylene glycol) (PEG) to develop a millimeter-thick brain-like tissue model based on the co-culture of neurons and glial cells. To set up a physiological model, we started from single cultures of primary mouse microglial cells, cortical neurons and astrocytes and compared two culture conditions: (a) a layered-based condition, where a layer of hydrogel covers the cells attached to the microplate; and (b) an embedded-based condition, where the cells are evenly distributed in the polymer solutions during hydrogel preparation. For both conditions, we recorded cell growth and survival up to 21 days, cell morphology and distribution, and synapse formation in neuronal networks. After selecting the most suitable condition, we proposed a more advanced model, co-culturing neurons and glial cells in the embedded-based condition.
Materials
For hydrogel preparation, we purchased reagents from Sigma-Aldrich (St. Louis, MO, USA). We obtained reagents for cell culture from Thermo Fisher Scientific (Waltham, MA, USA) and plasticware from Corning® (Corning, NY, USA).
Experimental procedures
Hydrogel preparation
For the development of 3D models, we cultured brain cells (as single cultures or co-cultures) in semi-IPNs based on COLL and HA (Mw=1・105 Da, Altergon Italia, Morra De Sanctis, Italy) or COLL and PEG. We used PEG with two molecular weights (Mw): 1.945 kDa (referred to as PEG2000) and 3.270 kDa (referred to as PEG3350).
For all the formulations, we diluted 8 parts (v/v) type I COLL solution (3 mg/mL) from bovine skin with 1 part (v/v) NaOH 0.1 N and 1 part (v/v) 10x phosphate-buffered saline solution (PBS). For COLL-HA gels, we mixed 1 part (v/v) COLL solution with 1 part (v/v) HA solution (5 mg/mL in water, autoclaved at 121 °C for 15 min or with an equivalent time F0 equal to 13). For COLL-PEG gels, we mixed 3 parts (v/v) COLL solution with 1 part (v/v) PEG solution (2.4 mg/mL in saline, autoclaved). To promote COLL fibrillogenesis, we incubated the polymer solutions at 37 °C for about 1.5 h.
To select the most suitable conditions to establish a brain-like tissue model based on the co-culture of neurons and glial cells, we plated the cells and covered with a 1.5 mm-thick hydrogel layer (layered condition) or we mixed 1 part (v/v) cell suspension with 9 parts (v/v) polymer solutions, and plated 1.5 mm-thick samples (embedded condition).
Primary brain cell cultures
Primary cells were harvested from CD-1 mice (Charles River Laboratories, Calco, Italy). We harvested microglia from 2 to 3 days postnatal mice, and cortical neurons and astrocytes from 2-4 days postnatal mice. We cultured the cells at 37 °C, 5% CO2.
3D microglial cells
We prepared microglial cultures according to previous studies.36,37 We removed the meninges and dissected a slice of cortex, minced it, digested it with 0.25% trypsin supplemented with 0.01% DNase I and pipetted it dropwise onto a 70-μm filter. We suspended the cells in Dulbecco’s modified eagle medium/Nutrient mixture F-12 Ham (DMEM/F-12), supplemented with 10% fetal bovine serum (FBS), 100 U/mL gentamicin, 100 U/mL penicillin and 100 µg/mL streptomycin, and plated them in flasks. When the cells were confluent (after about 15 days), we incubated them with 0.25% trypsin for 1.5 h, shaking the flasks every 30 min. We washed the adherent microglial cells twice with medium, detached them with 0.25% trypsin-0.02% EDTA, centrifuged them and suspended them in medium.
For layered conditions, we plated 63,000 cells/cm2 and covered them with a 1.5 mm-thick hydrogel layer. As controls, we plated 63,000 cells/cm2 on poly-L-lysine coated microplates. For embedded conditions, we mixed 158,000 cells/cm2 with the polymer solutions and plated 1.5 mm-thick samples.
For both conditions, after COLL fibrillogenesis we added culture medium and changed it every 2 days.
3D cortical neurons
We harvested primary cortical neurons according to Restelli et al. 38 Briefly, we removed the meninges, dissected small slices of cortex and incubated them at 34 °C for 30 min in dissociation medium supplemented with 20 U/mL papain. We added 0.5 mg/mL trypsin inhibitor and dissociated the tissue with a Pasteur pipette. We suspended the cells in Neurobasal medium supplemented with 2% B27, 2 mM L-glutamine, 100 U/mL penicillin and 100 µg/mL streptomycin.
For layered conditions, we plated 242,000 neurons/cm2, incubated them for 2h at 37 °C, and covered them with a 1.5 mm-thick hydrogel layer. As controls, we plated 242,000 cells/cm2 on poly-D-lysine coated microplates. For embedded conditions, we mixed 315,000 cells/cm2 with the polymer solutions and plated samples of 1.5 mm-thick. For both conditions, after COLL fibrillogenesis we added culture medium and refreshed it (50% v/v) every 2-3 days.
3D astrocytes
We dissected a part of the cerebral cortex, removed the meninges and collected the tissue in Hanks’ balanced salt solution (HBSS) without calcium chloride and magnesium, supplemented with 2 mM Hepes, 100 U/mL penicillin and 100 µg/mL streptomycin. We incubated it in 0.25% trypsin-0.02% EDTA for 20 min, added complete medium (Minimum Essential medium supplemented with 10% FBS, 6 g/L D-glucose, 100 U/mL penicillin and 100 µg/mL streptomycin) and dissociated it mechanically with a Pasteur pipette. We pipetted it dropwise onto a 40-µm nylon filter, centrifuged it, suspended it in complete medium and plated it in poly-L-lysine coated flasks.
When the mixed primary glial culture was confluent (after about 20 days), we shook the flasks overnight (220 rpm) and changed the medium. To obtain a purified astrocyte culture, the same or the next day we added L-leucine methyl ester (60 mM) to each flask for 90 min to inhibit microglial growth. Then, we washed the flasks with PBS and added fresh medium. The next day, we prepared the samples.
For layered conditions, we plated 52,500 cells/cm2 and covered them with a 1.5 mm-thick hydrogel layer. As controls, we plated 52,500 cells/cm2 on poly-L-lysine coated microplates. For embedded conditions, we mixed 105,000 cells/cm2 with the polymer solutions and plated 1.5 mm-thick samples. For both conditions, after COLL fibrillogenesis we added culture medium and changed it every 2-3 days.
3D cortical neurons and glial cells: embedded co-culture
We harvested the cells as described, but at least one day after shaking, astrocytes were cultured in complete medium for cortical neurons, and to preserve the microglial cells, L-leucine methyl ester was not added. We mixed 32,000 glial cells/cm2 and 315,000 cortical neurons/cm2 with the polymer solutions and plated 1.5 mm-thick samples. As controls, we co-cultured 13,200 glial cells/cm2 and 242,000 neurons/cm2 on poly-D-lysine coated microplates. After COLL fibrillogenesis, we added culture medium for cortical neurons and refreshed it (50% v/v) every 3 days.
Cell metabolic activity
To assess the ability of hydrogel matrices to support brain cell survival, we examined cell metabolic activity by MTS assay (Promega, Madison, WI, USA). This was done on days 1, 2 and 5 for microglial cells; on days 7, 14 and 18 for cortical neurons in layered conditions; and on days 7, 14 and 21 for the other conditions. We incubated the samples for 3 h with culture medium supplemented with 10% MTS, then measured their absorbance at 490 nm. We used 1.5-mm thick cell-free gels as control. We ran the assay on at least five samples/group for single cultures and ten samples/group for the co-culture.
Immunocytochemistry and confocal imaging
Immunocytochemistry was done to observe brain cell morphology and spatial distribution at the time points when we measured the highest cell metabolic activity for 2D controls (on day 2 for microglial cells, and on day 14 for cortical neurons, astrocytes and the co-culture).
We removed the culture medium, washed the samples with PBS and fixed them with paraformaldehyde solution (PFA, 4% in PBS), for 24 h. We washed them with Tris-buffered saline supplemented with 0.1% Tween 20 (TBS-T), and blocked them (8–9 h, 4°C) in TBS-T with 4% normal goat serum (NGS), 1 mg/mL bovine serum albumin (BSA), Tris 50 mM, 0.1% gelatin and 0.3 M glycine. After washing the samples with TBS-T, we incubated them (20 h, 4 °C) with primary antibodies, then washed and incubated (10 h, 4 °C) with secondary antibodies. We stained cell nuclei with 0.02 mM Hoechst 33342 (Thermo Fisher Scientific) for observation with a confocal microscope (FV10i, Olympus, Tokyo, Japan). We stained microglial cells with anti-CD11b (1:8000, Biotechne, Minneapolis, MN, USA), and anti-CD68, a pan-macrophage marker (1:200, Bio-Rad Laboratories, Hercules, CA, USA), cortical neurons with anti-neuron-specific β-tubulin III (Tuj-1, 1:500, Sigma-Aldrich), and astrocytes with anti-glial fibrillary acidic protein (GFAP, 1:3000, Millipore, Burlington, MA, USA). As secondary antibodies (1:500, Thermo Fisher Scientific) we used anti-mouse IgG Alexa Fluor® 488, anti-mouse IgG Alexa Fluor® 647, and anti-rabbit IgG Alexa Fluor® 594. As controls, we examined cells in 2D conditions. Only for single cultures of microglial cells, on day 2 we investigated whether hydrogel presence elicited microglial activation by comparison with control cells previously incubated for 24 h with lipopolysaccharide (LPS, from Escherichia coli O111:B4, 1 ug/ml, Sigma-Aldrich), a pro-inflammatory stimulus.
Western Blotting
To assess neuronal function, on days 7 and 14 we investigated synapse formation in single cultures of cortical neurons and in the co-culture. To examine the presence of astrocytes, we investigated GFAP expression in the co-culture at the same time points.
We removed the culture medium, washed the samples with PBS, added lysis buffer (with 1% protease inhibitor cocktail), incubated them for 1 h at 4 °C and lysed them with a cell scraper. We vortexed them for about 1 min, then centrifuged them at 4 °C for 5 min at maximum speed and collected the supernatants. To evaluate the effect of 3D embedded conditions on the basal levels of protein expression, we also examined controls in 2D conditions. In view of a possible interference from COLL, protein content was evaluated only for controls, by the bicinchoninic acid assay (BCA, Pierce™, Thermo Fisher). We separated protein extracts by SDS-PAGE and transferred them to a nitrocellulose membrane (Bio-Rad Laboratories). We incubated the membranes overnight at 4 °C with anti-postsynaptic density protein 95 (PSD-95, 1:5000, NeuroMab, Davis, CA, USA), anti-synaptophysin (a presynaptic protein, 1:5000, Synaptic System GmbH, Goettingen, Germany), anti-N-Methyl-D-Aspartate Receptor Subunit NR1 (NR-1, 1:1000, Synaptic System GmbH), anti-GFAP (1:10000), anti-Tuj-1 (1:5000) or glyceraldehyde 3-phosphate dehydrogenase (GADPH, 1:2000, Millipore). Then, we incubated the membranes with the secondary antibodies conjugated to the enzyme horseradish peroxidase. We covered the membranes with luminol solution (EMD Millipore), developed the immunoreactive bands with Firereader V10 PLUS 26M Imaging system (Uvitec Ltd, Cambridge, UK) and quantified them with Image Lab software. The analysis was done in triplicate.
Statistics
The results were reported as mean ± standard deviation (SD) and analyzed with GraphPad Prism® software (GraphPad Software Inc, release 8.0). Two-way analysis of variance (ANOVA) was followed by Tukey’s multiple comparisons test for comparisons of the groups and time frames. We considered p < 0.05 significant.
Results
3D microglial cells
In the absence of supporting cells like astrocytes, single cultures of primary microglial cells are viable for a very short time 39 and usually tested 48 to 72 h after plating. We therefore limited the evaluation of their metabolic activity to 5 days. However, this was sufficient to appreciate the benefits of hydrogels for extending their culture time.
In layered conditions, metabolic activity (upper panel, Figure 1(a)) was constant (p > 0.05) from day 1 to 2 for cells covered with COLL-HA gels, and it decreased with PEG-based gels. From day 2 to 5, it increased for all the matrices (p <0.0001 for all comparisons). The significant increase on day 5 was due to the loss of viability of 2D controls (almost total absence of cells) and the general slowing of cell growth observed for the hydrogels. On day 2, confocal images (upper panel, Figure 1(b)) confirmed uniform cell distribution and nuclei staining indicated that cell number was comparable in all conditions. CD11b staining showed a larger number of branches in the presence of the hydrogels, suggesting the induction of microglial activation. PEG-based matrices showed greater microglial activation than COLL-HA ones, but still lower than positive controls treated with LPS.

Primary microglial cells in layered and embedded conditions. Layered conditions: (a) Cell metabolic activity over time for microglial cells covered with a 1.5 mm-thick layer of COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. The results are mean ± SD with respect to 2D controls (CTRL), 5 replicates/condition. (b) Representative images of microglia immunostained for CD11b (red) and cell nuclei stained with Hoechst 33342 (blue) on day 2. As a reference, cells grown in 2D conditions and incubated with LPS (CTRL + LPS) were also immunostained. Scale bar: 20 μm.
In embedded conditions, cell metabolic activity (lower panel, Figure 1(a)) was constant (p > 0.05) over time for COLL-HA gels, while for PEG-based semi-IPNs it decreased from day 1 to 2, and then remained constant (p > 0.05). CD11b staining (lower panel, Figure 1(b), first set of images) indicated that a few microglia presented a more branched morphology than controls. This suggests that microglial activation is almost absent in embedded conditions. We also assessed microglial phagocytic activity by CD68 staining (red in the second set of images in Figure 1(b)). In COLL-HA and COLL-PEG2000 gels, we detected immunoreactivity in the cytoplasm of some microglia, indicating that cell morphology elicited an immune response. For controls and COLL-PEG3350 gels, activated cells were almost absent.
3D cortical neurons: Metabolic activity and immunocytochemistry
Experimentally, neuronal cultures are usually employed within 14 days. However, extending this observation time may be important in the view of developing an OOC-based platform for prolonged cell culture and improving the in vitro recapitulation of pathological mechanisms. We therefore extended this target time up to 21 days.
In layered conditions (upper panel, Figure 2(a)), the gels reduced cell metabolic activity compared to 2D controls. Among the matrices, at all the time points PEG-based hydrogels performed best. On day 18, neurons covered with COLL-PEG3350 showed the greatest metabolic activity (p < 0.0001), while COLL-HA and COLL-PEG2000 semi-IPNs gave comparable results (p < 0.05). On day 14, for 2D controls and layered cells, Tuj-1 staining (upper panel, Figure 2(b)) indicated the presence of articulated neuronal networks and uniform spatial distribution of neuronal extensions, with no aggregated branches. Control neurons had more complex networks. However, neurons covered with PEG-based gels showed more numerous extensions than controls and neurons in COLL-HA semi-IPNs.

Primary cortical neurons in layered and embedded conditions. Layered conditions: (a) Cell metabolic activity over time for cortical neurons covered with a 1.5 mm-thick layer of COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. The results are mean ± SD with respect to 2D controls (CTRL), 8 replicates/condition. (b) Representative images of neurons immunostained for β-tubulin III (Tuj-1, green) and cell nuclei stained with Hoechst 33342 (blue) on day 14. Scale bar: 50 μm.
Starting from these promising results, we extended the culture time to 21 days for the embedded condition (lower panel, Figure 2(a)). At all the time points, cell metabolic activity was lower for COLL-HA semi-IPNs. Differences between PEG-based gels were found only on day 21, with COLL-PEG2000 performing best (p < 0.0001). On day 14 (lower panel, Figure 2(b)), 2D controls showed dense neuronal networks. Similar networks were observed in PEG-based semi-IPNs, where neuronal extensions penetrated the matrices and spread in all directions. For COLL-HA gels, MTS assay indicated viable neurons, but Tuj-1 staining showed the almost complete absence of neuronal extensions. DNA staining indicated smaller nuclei than in the other semi-IPNs, suggesting possible cell suffering. These results indicate that the embedded condition promotes the formation of more complex neuronal networks than the layered one. The embedded-based model represents a more physiologically relevant condition and allows cells to benefit from a larger surface area for growth. 40
3D cortical neurons: Synapse formation
To obtain dense, mature neuronal networks in vitro, generally about 10 days are required. 41 We therefore studied the expression of PSD-95, synaptophysin, and NR-1 on days 7 and 14. Since it is a neuron-specific marker and it is not influenced by cell proliferation, we investigated synapse formation using β-tubulin III as the housekeeping protein. As stress or 3D culture conditions can influence cytoskeleton architecture and thus β-tubulin III expression, 42 before quantification we performed Western blot analysis using GADPH as the housekeeping protein (data not shown).
In layered conditions (upper panel, Figure 3(a)–(d)), the levels of PSD-95 rose from day 7 to 14 for PEG-based gels (p < 0.01), but decreased in the presence of COLL-HA (p < 0.01). In these matrices, the levels of synaptophysin also decreased (p < 0.0001). No differences in the levels of NR-1 were observed for the hydrogels.

Immunoblots on primary cortical neurons in layered and embedded conditions. Layered conditions: Western blot quantification on days 7 and 14 to detect (a) PSD-95, (b) Synaptophysin, (c) NR-1, in cortical neurons covered with a 1.5 mm-thick layer of COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. (d) Representative immunoblots used for the quantifications.
In contrast, in embedded conditions (lower panel, Figure 3(a)–(d)), the levels of PSD-95 increased for PEG-based gels from day 7 to 14, but it was not significant (p > 0.05). Similarly, for all the matrices the levels of synaptophysin did not change over time (p > 0.05). On the contrary, the levels of NR-1 rose for COLL-HA samples (p < 0.001).
3D astrocytes
For astrocytes in layered conditions (upper panel, Figure 4(a)), cell metabolic activity was lower than for 2D controls. For all the matrices, cell metabolic activity increased (p < 0.0001) between days 7 and 14, but the worst performances were for COLL-HA semi-IPNs. On day 21, metabolic activity was greatest for astrocytes layered with PEG-based gels (p < 0.0001). On day 14, confocal images (upper panel, Figure 4(b)) showed uniform cell distribution. For astrocytes covered with COLL-HA gels, nuclear staining indicated a cell number comparable to the other conditions, but GFAP staining suggested that astrocytes had a limited, undeveloped morphology compared to their counterparts covered by PEG-based gels.

Primary astrocytes in layered and embedded conditions. Layered conditions: (a) Cell metabolic activity over time for astrocytes covered with a 1.5 mm-thick layer of COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. The results are mean ± SD with respect to 2D controls (CTRL), 5 replicates/condition. (b) Representative images of astrocytes immunostained for GFAP (green) and cell nuclei stained with Hoechst 33342 (blue) on day 14. Scale bar: 50 μm.
In embedded conditions (lower panel, Figure 4(a)), cell metabolic activity was greatest for COLL-HA semi-IPNs (p < 0.0001) and no differences were found between PEG-based gels. This suggests slower astrocyte proliferation in PEG-based matrices. Like in layered conditions, on day 14 confocal images (lower panel, Figure 4(b)) showed uniform cell distribution within the gels. In COLL-HA semi-IPNs, nuclei staining indicated a higher cell number than in 2D controls and PEG-based matrices, but GFAP staining suggested that COLL-HA gels disadvantaged astrocytic branching. There were some differences between astrocyte morphology in controls and PEG-based gels, but growth in 3D matrices seemed slower than in 2D conditions.
3D neurons and glial cells: Co-culture (metabolic activity and immunocytochemistry)
The previous results indicated that the embedded-based model promotes the formation of more complex neuronal networks and considerably reduces microglial activation. We therefore selected this condition to develop a 3D brain-like tissue model based on the co-culture of cortical neurons and glial cells.
In COLL-HA gels, cell metabolic activity (Figure 5(a)) was constant from day 7 to 14 (p > 0.05), then it decreased (p < 0.01). In PEG-based matrices, it decreased from day 7 to 14 (p < 0.0001), then it rose (p < 0.05 for COLL-PEG2000; p < 0.001 for COLL-PEG3350). On day 21, COLL-PEG3350 semi-IPNs performed best.
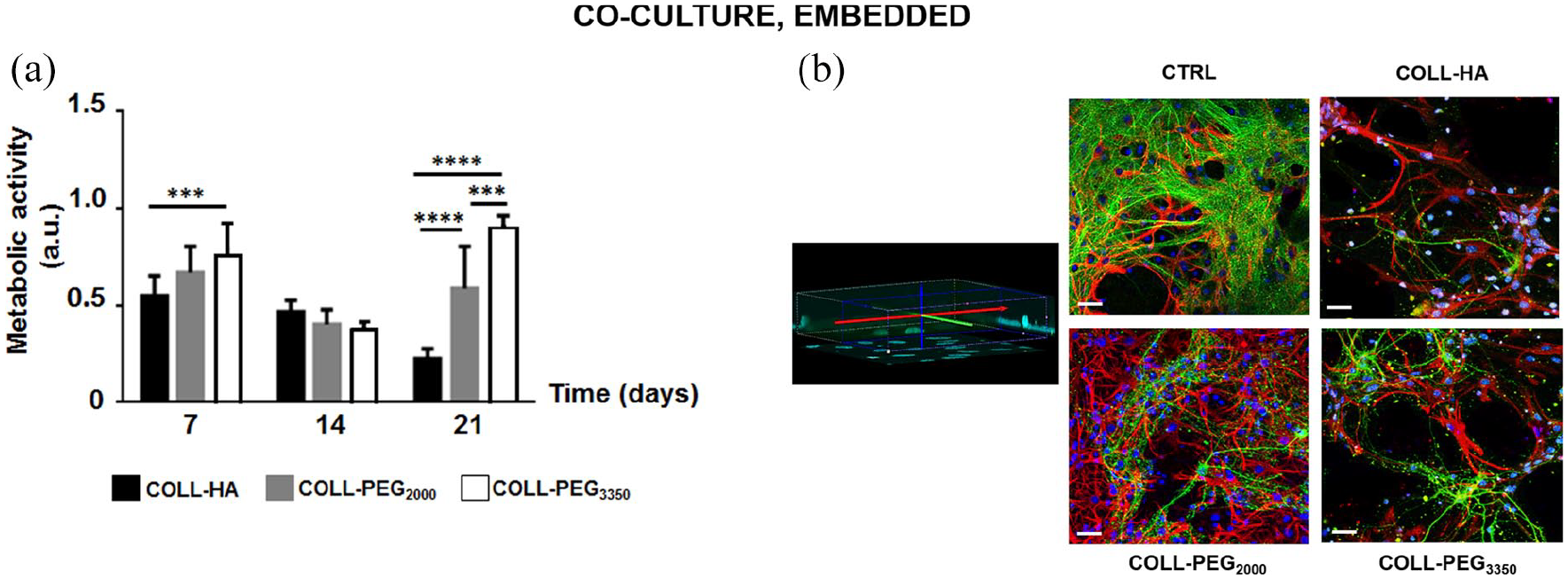
Co-cultures of cortical neurons and glial cells. (a) Cell metabolic activity over time for cortical neurons and glial cells embedded in 1.5 mm-thick COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. The results are mean ± SD, 10 replicates/condition. The results were analyzed with ordinary two-way ANOVA followed by Tukey’s multiple comparisons test. (b) Representative images of embedded neurons immunostained for β-tubulin III (Tuj-1, green), astrocytes immunostained for GFAP (red) and cell nuclei stained with Hoechst 33342 (blue) on day 14. The morphology of CTRL was reported for comparison. Some nuclei whose cell body was not reactive either to Tuj-1 or GFAP are present, likely belonging to microglial cells. Scale bar: 20 μm.
On day 14, confocal imaging showed the presence of complex networks (Figure 5(b)), where glial cells and neurons can communicate. GFAP staining indicated that astrocytes supported neuronal cells, while Tuj-1 staining showed that neural networks are denser than in single cultures. However, spatial organization was more complex in COLL-PEG2000 gels. In agreement with nuclear staining, in COLL-HA and COLL-PEG3350 matrices, Tuj-1 staining revealed less dense and structured neuronal networks, while GFAP staining showed that glial cells were only partially dedicated to neuronal support.
3D neurons and glial cells: Co-culture (synapse formation)
When co-culturing neurons and glial cells, we measured the levels of synaptic markers in the semi-IPNs at 7 and 14 days (Figure 6(a)–(d)). These suggest that the selected culture conditions permit the maintenance of the key biochemical parameters of the embedded cell populations for a considerable time. In most cases (e.g. PSD-95 in COLL-PEG3350, synaptophysin in COLL-PEG2000 and NR-1 in COLL-HA gels), we confirmed the previous results for single cultures of cortical neurons in embedded conditions. Furthermore, the changes in expression levels were greater in co-culture conditions than in single cultures. However, for COLL-HA gels protein content was lower than for PEG-based gels (Figure 6(e)). This agrees with previous results from cell metabolic activity (Figure 5(a)), but suggests a poor quality of the culture. In COLL-PEG3350, β-tubulin III expression was low on day 7, suggesting slower neuronal maturation than in COLL-PEG2000 matrices. However, it rose significantly on day 14. In COLL-PEG2000 gels, β-tubulin III increased slightly between days 7 and 14, indicating that these semi-IPNs are more promising to reach the target culture time of 21 days.

Immunoblots on co-cultures of cortical neurons and glial cells. Western blot quantification on days 7 and 14 to detect (a) PSD-95, (b) Synaptophysin, (c) NR-1, (d) GFAP, in co-cultures of cortical neurons and glial cells embedded in 1.5 mm-thick COLL-HA, COLL-PEG2000 or COLL-PEG3350 gels. The results (mean ± SD, 3 replicates/condition) are also reported for 2D controls (CTRL) and normalized to β-tubulin III (Tuj-1). They were analyzed with ordinary two-way ANOVA followed by Tukey’s multiple comparisons test. (e) Representative immunoblots used for quantification. For CTRL, we loaded 20 μg total protein, and for each hydrogel, we loaded 20 μL total sample.
To investigate the balance between neurons and glial cells over time, we measured the expression levels of GFAP from days 7 to 14 and noted an increase (although not significant) only for COLL-PEG3350 matrices (Figure 6(d)). These results confirm that this matrix promotes astrocyte proliferation, but in the time window examined glial cells do not take over the neuronal network.
Discussion
The bidirectional communication between intestinal microbiota and the brain, referred to as MGBA, has attracted growing interest also for its clinical potential, such as exploiting microbiota-based therapeutics as new strategies to manage neurological diseases and behavioral functions. Several challenges remain before clinical translation, 43 but a reliable OOC platform recapitulating in vitro the main players of the MGBA may offer an effective tool to clarify the impact of intestinal microbiota on brain functions, in physiological and pathological conditions. In this perspective, developing an OOC-based 3D system modeling brain tissue is particularly challenging. 4
Starting from the scalability of the thickness of COLL-based semi-IPNs to the millimeter scale and their suitability for dynamic culture in a microfluidic, optically accessible OOC device, 5 we set up a novel 3D brain-like tissue model potentially suitable for dynamic culture by coupling 1.5 mm-thick COLL-based semi-IPNs to the co-culture of cortical neurons and glial cells. Semi-IPNs have the advantage of easy tunability of their final properties, 35 a key point when aiming at co-culturing different cell types, including neural cells.
Initially, we investigated the biocompatibility of the COLL-based semi-IPNs with primary mouse microglia, neurons and astrocytes as single cultures. Compared to Matrigel, widely exploited as a scaffold material for neural cells,39,44,45 COLL has a more defined composition and has also been successfully applied for brain cultures.46,47
Since different 3D culture conditions are reported31,32 and their advantages depend on the application, 48 we considered both a layered-based model, where cells are covered by the hydrogel, and an embedded-based model, where cells are uniformly dispersed in the 3D volume. In both conditions, neurons maintained their phenotype and showed more interconnected networks than in 2D cultures. However, more complex neuronal networks and the absence of microglial activation due to the hydrogel were found in the embedded condition, indicating that it is preferable. We therefore co-cultured neurons and glial cells only in the embedded-based situation. For both embedded neurons and microglial cells, PEG-based gels performed better. In COLL-HA semi-IPNs, neuron viability was reduced, as indicated by the failure to emit extensions and build a complex network in confocal imaging and the low expression of synaptic proteins. However, astrocytes proliferated more in COLL-HA semi-IPNs. This is a key point with a view to co-culturing neurons and glial cells, since excessive astrocyte proliferation could hinder neuronal survival. With this in mind, PEG-based formulations may be the best-balanced solution also for astrocyte culture.
The importance of astrocyte-neuron ratio was reported by Fang and colleagues, who showed that the spatial relationships between neurons and astrocytes affect neuronal growth and functions. 48 To mimic the ratio between the number of glial cells and neurons in the natural brain cortex, 49 they co-cultured astrocytes and neurons on a 1:2 ratio for 14 days and showed that neurons benefited from astrocytes when these were confined by the neurons. In contrast, in the embedded condition, astrocytes over-expanded and hindered neuronal growth. 49 To avoid this, in the co-culture we used fewer glial cells than in single cultures and optimized a ratio of 1:10 between glial cells and neurons. This ratio was also considered in other studies, for instance with induced-pluripotent stem cells (iPSC). 50
An important point when developing 3D brain cell models is to achieve sufficiently long-lasting viability to allow cell maturation and full differentiation. In our models, MTS assay confirmed cell viability up to 21 days, with a better performance for PEG-based gels. Although metabolic activity was greater for cells in COLL-PEG3350, we suggest COLL-PEG2000 as the best option for the full 3D model because of the better structured neuron/glia networks. For this reason, it appeared as the most promising candidate for a 3D brain-like tissue model and would also be suitable to study the impact of intestinal microbiota on brain function in an OOC device. However, a further characterization of the model is mandatory, in particular by performing microglia immunostaining with suitable antibodies (e.g., CD11b or Iba1).
From the cell model point of view, primary cells are more physiological than continuous cell lines and have some advantages in terms of costs and reproducibility compared to other systems, such as those based on stem cells. The literature reports several examples of 3D brain-like tissue models exploiting stem cells, where cell differentiation is achieved with differentiation media or based on a combination of hydrogel formulation and final mechanical properties.51–53 Recent works have focused on iPSC, which can be differentiated into neural progenitor cells, and then various CNS-related cell phenotypes. They are excellent candidates for personalized medicine 54 and disease modeling.55–57 For instance, starting from the work by Choi et al., 40 Park et al. employed iPSC to successfully reproduce dynamic neural-glial interactions in a microfluidic device and recapitulate key features of Alzheimer’s disease, such as beta-amyloid accumulation, phosphorylated tau aggregation and neuroinflammation. 57
However, the differentiation of iPSC-derived progenitor or precursor cells is hard to control, 58 and iPSC-derived neuronal cells are still not suitable for time-consuming optimization studies such as the one presented here, because they are expensive. Furthermore, although the differentiation protocols have been optimized to confirm the nature of post-differentiation cells in terms of functionality and morphology, the technologies still need improvements to boost reproducibility. 59 In the future, the use of commercial iPSC-derived microglial cells will extend the limited culture time of their primary counterparts, thus overcoming a current limitation of our study and improving the predictability of the 3D systems. Abud et al. described the generation of highly pure iPSC-derived human microglial-like cells similar to both fetal and adult microglia to study their functions in physiological and pathological conditions, like Alzheimer’s disease. 60
When recapitulating brain physiology at the molecular and functional levels, key points are the evaluation of synaptic proteins, suggesting the maturation of the neural network, and the recording of electrophysiological activity. For electroactive cells in 2D conditions, the recording of electrophysiological activity is widely consolidated,32,61 while the development and optimization of electrodes and stimulation/recording devices for cells in 3D conditions is an active research field. Since the possibility of stimulating or recording relies on close proximity to the electrodes, the thickness of the culture system is important for the feasibility and reliability of the acquisition. Commercial electrodes penetrate 50 to 100 µm into the tissue, but electrophysiological measurements are reported for thicker systems. Frega et al. stacked 5-8 layers of microbeads with diameter about 40 µm and hippocampal neurons, 62 while recently Soscia et al. developed and validated an integrated 3D microelectrode array 63 for applications with 750 to 1000 µm-thick cell-loaded hydrogels. For our goals, we preliminarily investigated our 3D brain-like models using a standard electrophysiology apparatus with a patch-clamp setup, but the background noise was too high to distinguish signals from the embedded cells. Thus, further experiments and technical improvements are still needed.
Conclusions
We developed an in vitro physiological brain-like tissue model based on 1.5 mm-thick hydrogels and primary neuronal cells, also co-cultured, in both layered and embedded conditions. It provides a 3D model for setting up a brain compartment to be inserted into a complex OOC platform recapitulating brain functional relationship with peripheral body districts, as in the MGBA, in pathological and physiological conditions.
Future developments will focus on exploiting these results to develop a 3D, iPSC-based, brain-like tissue model, also for investigating Alzheimer’s disease.
Footnotes
Acknowledgements
The authors are very grateful to Elena Restelli and Paola Fabbrizio (Istituto di Ricerche Farmacologiche Mario Negri IRCSS, Milan) for useful suggestions about cell culturing. We also thank Felice Volpe (Altergon Italia srl, Morra De Sanctis, Italy; currently at IBSA Institut Biochimique SA. Lugano, Switzerland) for suggestions about HA handling and processing and Dr. Judith Baggott for her skilled English editing.
Author contributions
IR, DA and CG conceived the study. IR carried out the experiments with support from MT for hydrogel preparation. IR and MT wrote the manuscript. GF made the equipment at the Dept. Neuroscience, Istituto di Ricerche Farmacologiche Mario Negri IRCCS, available for the experiments. DA and CG supervised the present manuscript and the related project activities. All authors discussed the results and approved the final manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was funded by the European Research Council (ERC) under the European Union’s Horizon 2020 research and innovation program (G.A. 724734-MINERVA) to CG. The results reflect only the authors’ views and the Agency is not responsible for any use that may be made of the information contained.