
Abstract
Under appropriate culture conditions, mesenchymal stem cells (MSC), also called more properly multipotent mesenchymal stromal cells (MMSC), can be induced toward differentiation into different cell lineages. In order to guide stem cell fate within an environment resembling the stem cell niche, different biomaterials are being developed. In the present study, we used silk fibroin (SF) as a biomaterial supporting the growth of MMSC and studied its effect on chondrogenesis of canine adipose–derived MMSC (cADMMSC). Adipose tissue was collected from nine privately owned dogs. MMSC were cultured on SF films and SF scaffolds in a standard cell culture medium. Cell morphology was evaluated by scanning electron microscopy (SEM). Chondrogenic differentiation was evaluated by alcian blue staining and mRNA expression of collagen type 1, collagen type 2, Sox9, and Aggrecan genes. cADMMSC cultured on SF films and SF scaffolds stained positive using alcian blue. SEM images revealed nodule-like structures with matrix vesicles and fibers resembling chondrogenic nodules. Gene expression of chondrogenic markers Sox9 and Aggrecan were statistically significantly upregulated in cADMMSC cultured on SF films in comparison to negative control cADMMSC. This result suggests that chondrogenesis of cADMMSC could occur when cells were grown on SF films in a standard cell culture medium without specific culture conditions, which were previously considered necessary for induction of chondrogenic differentiation.
Keywords
Introduction
Mesenchymal stem cells (MSC), more properly called multipotent mesenchymal stromal cells (MMSC), have received significant interest for their potential use in regenerative therapy in human and veterinary medicine due to their immunosuppressive and multilineage differentiation capabilities. 1 There are several established protocols for induction of MMSC differentiation in vitro, which are optimized for conventional culturing of MMSC in two-dimensional (2D) cell culture system with polystyrene vessels. Under appropriate culture conditions, differentiation of MMSC can be induced toward adipocytic, osteocytic, and chondrocytic lineages.2,3 Chondrogenesis can be induced with specific culture conditions such as high cell density, or by induction with different hormones and growth factors, particularly TFG-β.4,5 However, there are also several reports describing spontaneous differentiation of MMSC toward different lineages.6–9
The key factors responsible for MMSC differentiation are not yet known. Current stem cell research is trying to understand the mechanisms needed for guiding stem cell fate in a desired manner. 10 Traditional culturing of MMSC on 2D surfaces, such as cell culture polystyrene is thus being replaced by three-dimensional (3D) cell culture techniques. In vivo cells reside in a very complex 3D environment, which plays an essential role in determining stem cell fate and regulating their self-renewal and differentiation. In vitro, however, the culture conditions are very different, especially in classical 2D systems, and this raises the questions how much the cells cultured in vitro resemble cells inside the organisms. Therefore, there is a need for the development of novel biomaterials, which could provide appropriate physiologically relevant biochemical and mechanical signals in order to guide stem cell fate 10 and to restore tissue functions. 11 It has been demonstrated that mechanical cues,12–14 surface chemistry,15–17 and physical properties18–20 of biomaterials play key roles in the regulation of stem cell differentiation.
Different biomaterials such as polyetherurethane and poly(ether imide), 21 polyethylene glycol-linked multi-walled carbon nanotube films, 22 reduced graphene oxide-coated hydroxyapatite composites, 23 poly-lactic-co-glycolic acid nano-fiber scaffold, 24 and electrospun nanofibrous scaffolds 25 have been shown to promote differentiation of MMSC into adipogenic, myogenic, osteogenic, and chondrogenic lineages. However, the mechanisms regulating these differentiation processes are not yet understood. Until now, there is no universal biomaterial that could meet all the requirements for different tissues with specific physical and mechanical properties. One of the promising biomaterials to be used in tissue engineering is silk fibroin (SF), derived from the silkworm Bombyx mori. It is biocompatible, has suitable mechanical properties, and is produced in bulk in the textile industry. 26 In comparison to other polymers used for tissue engineering, SF provides a remarkable combination of strength, toughness, and elasticity that are assigned to its crystallinity, hydrogen bonding, and numerous small β-sheet crystals. 27 Moreover, as a natural biopolymer, it degrades to non-toxic and neutral degradation products (amino acids and peptides), whereas the degradation is slow and controllable. Although it is a natural biopolymer, it could be sterilized using common sterilization techniques, it has high thermal stability, and the processing of materials from silk can be aqueous-based. 28 Furthermore, SF can be fabricated into different materials such as hydrogels, tubes, sponges, composites, fibers, microspheres, and films that could be used in tissue engineering. 29 Various modifications of SF properties can aid in stem cell proliferation and differentiation potential,30–35 although it has not been reported before that SF promotes differentiation of MMSC. In this study, we demonstrate that SF guide canine adipose–derived MMSC (cADMMSC) toward presumed chondrogenic differentiation.
Materials and methods
Adipose tissue collection
Subcutaneous adipose tissue was collected from nine privately owned dogs. Adipose tissue was individually collected during routine clinically indicated surgery at Small Animal Clinic of the Faculty of Veterinary Medicine in Ljubljana. All samples were randomly assigned to different experimental groups. All owners agreed with the collection of tissue and signed an informed consent. Since study was conducted on client-owned animals undergoing routine clinical procedure with owner’s approval to collect small piece of adipose tissue, no approval of ethical committee was needed according to Slovenian legislation and official opinion from The Administration of Republic of Slovenia for Food Safety, Veterinary and Plant protection, responsible for issuing ethical permits for animal experiments.
Isolation and culture of cADMMSC
Immediately after collection, adipose tissue was washed with Dulbecco’s Phosphate Buffered Saline (DPBS, Gibco, USA) and cut with a scalpel into small pieces. Adipose tissue was then incubated overnight at 37°C in Dulbecco-modified Eagle medium (DMEM, Gibco, USA) containing 0.1% collagenase type II (Sigma-Aldrich, DE). The digested tissue was centrifuged at 1600 r/min for 4 minutes, and the supernatant was discarded. Pellet of cells was resuspended in cell culture medium containing DMEM and 10% Fetal Bovine Serum (FBS, Gibco, USA). The cell suspension was plated into 6-well plates (TPP, Switzerland) and cultured at 37°C in a 5% CO2 incubator. Cell culture medium was changed every 2–3 days. After 80%–90% confluency was reached, cells were trypsinized and multiplied by seeding 104 cells per cm2 into a larger (T25) cell culture flask. Cells in cell culture were maintained up to the fourth passage. After a sufficient number of cells was reached, cells were used for assessing chondrogenic differentiation. All cells used in the experiments were from the second, third or fourth passage, and cells were cultured for 2 or 3 days in each passage. All experiments were repeated five times for positive and negative controls and six times for cells cultured on SF films (both 1 week and 2 weeks).
Multilineage differentiation potential of cADMMSC
Differentiation potential was assessed by inducing cADMMSC differentiation into adipocytes, osteocytes, and chondrocytes. For the adipogenic and osteogenic differentiation 4 × 104 cells were seeded in 12-well plates. When 90% confluency was reached, the cell culture medium was removed. Adipogenic (StemPro Adipogenesis Differen-tiation Kit, Gibco, USA) or osteogenic (StemPro Osteogenesis Differentiation Kit, Gibco, USA) medium was added and changed every 2–3 days. Cell culture medium was added to the wells that served as negative controls. Adipogenic differentiation was analyzed with Oil-red-O staining (Sigma-Aldrich, DE) after 21 days of culturing. Osteogenic differentiation was analyzed with Alizarin Red S staining (Sigma-Aldrich, DE) after 14 days of culturing, which is shorter than recommended for human cells, but during our preliminary studies, we have established that with canine cells, 21 days of culture is too long period (cells start to detach) while majority of cells stain positive with Alizarine red already after 14 days of culturing. For the chondrogenic differentiation, micromass cultures were generated by seeding 5 µL droplets of 4 × 104 cells in the center wells of 12-well plate. After cultivating micromass cultures for 6 hours under high humidity conditions, chondrogenic medium (StemPro Chondrogenesis Differentiation Kit, Gibco, USA) was added to culture vessels. Regular cell culture medium was added to the wells that served as negative controls. Micromass cultures were incubated in 37°C incubator with 5% CO2 and humid atmosphere. The medium was changed every 2–3 days. Chondrogenic differentiation was analyzed with alcian blue staining (Sigma-Aldrich, DE) after 14 days of culturing.
Preparation of SF films and scaffolds
SF films and scaffolds were prepared following the procedure described by Rockwood et al. 36 Briefly, Bombyx mori silk cocoons were cut in pieces and boiled for 30 minutes in 0.02 M solution of sodium carbonate (Na2CO3) to extract sericin. SF was rinsed in ultrapure water several times until the conductivity of water became constant and then dried overnight at 65°C. Degummed SF was dissolved in 9.3 M lithium bromide (LiBr) solution at 72°C for 3 hours and then subsequently dialyzed in a constant flow (0.4 L h−1) of ultra pure water at 4°C until its conductivity fell below 0.5 µS. The molecular weight cut off of dialysis tubing cellulose membrane was 12–14 kDa. To eliminate impurities, the prepared solution was centrifuged at 20,000 r/min for 20 minutes. The concentration of SF solution was determined by Bradford assay protocol 37 based on the color change of Coomassie Brilliant Blue G-250 using Bio-Rad protein assay (Bio-Rad laboratories, Hercules, CA). The SF solution was added to the Bradford reagent and incubated for 5 minutes. The absorbance was measured at 595 nm. Two different concentrations of SF solution were used for the preparation of scaffolds and films. The concentration of the prepared solution was on average 8 mg/mL and was used for the preparation of scaffolds, whereas a higher concentration of SF solution, 12.5 mg/mL, was used for the preparation of films. Higher concentration was achieved using centrifugation through centrifugal filter units (Amicon Ultra–4 centrifugal filter unit, Merck, Cork, IE).
SF films were prepared by casting 300 µL of the SF solution (12.5 mg/mL) into the wells of 12-well plates with a subsequent overnight air-drying. Films were then incubated in 70% ethanol for 10 minutes. In the last step, films were thoroughly washed with PBS.
SF scaffolds were prepared by adding 300 µL of SF solution (8 mg/mL) into the wells of 48-well plate. SF solution in well plates was then frozen in liquid nitrogen and lyophilized at −50°C for 72 hours to sublimate water and thus form porous scaffolds. After lyophilization, SF scaffolds were soaked in absolute ethanol overnight and then dried in a desiccator. Finally, scaffolds were thoroughly washed with PBS to remove any remaining ethanol.
SF scaffold characterization
Porosity and the pore size distribution of the SF scaffolds were determined using a Pascal series mercury intrusion porosimeter (Thermo Scientific). The surface tension and the contact angle of the mercury were set to 0.485 and 140 mN/m, respectively.
Wettability of the SF film was evaluated by measuring water droplet contact areas of the curve fitted to the droplet image on a dry and wet SF film using the Contact Angle Instrument (First Ten Ångstroms, Inc., USA, FTA1000 series). The measurement system consisted of a sample stage, vertically fitted Hamilton micro-syringe to place the water droplet on the sample and the camera mount–TV lens camera with Extension tube set 40 mm (Edmund optics, Japan). Images were captured and analyzed for contact areas using the FTA32 Video 2.0 software.
Cultivation of cADMMSC on SF films and SF scaffolds
After a sufficient number of cells was obtained, cells were cultured in four different ways:
(1) On SF films in cell culture medium for 7 and 14 days: 104 cells per cm2 were seeded onto 12-well plate with wells coated with SF films.
(2) On SF scaffolds in cell culture medium for 14 days: 9 × 5 µL droplets of 1 × 105 cells were seeded onto the bottom side of the SF scaffolds. During scaffold preparation, membrane-like portion of SF formed on the top of the scaffolds making the scaffold impassable for cells. Therefore, scaffolds were carefully lifted from the wells and turned upside down. Cells were then seeded onto the scaffolds.
(3) On a standard polystyrene surface in chondrogenic medium for 14 days: cells were cultured as described above for a multilineage differentiation potential.
(4) On a standard polystyrene surface in standard cell culture medium until 80%–90% confluency was reached.
Cell cultures were named accordingly (Table 1).
Name of the cell cultures, cell seeding surfaces and densities, culture media, and culture conditions used in the study.
cADMMSC, canine adipose–derived multipotent mesenchymal stromal cells; SF, silk fibroin.
Alcian blue and DAPI staining
For alcian blue staining of SF film cADMMSC, positive control cADMMSC, and negative control cADMMSC, medium was removed from culture vessels. Wells were rinsed once with DPBS and cells were fixed with 4% paraformaldehyde solution for 1 hour. Following fixation, wells were rinsed twice with DPBS and incubated overnight in 1% alcian blue stain (pH 2.5), prepared in 0.1 N HCl. Next day wells were rinsed three times with 0.1 N HCl followed by DPBS to neutralize the acidity. Wells were examined under the light microscope. For comparison of alcian blue stained presumed chondrogenic nodules from positive control cADMMSC and SF film cADMMSC, some presumably chondrogenic nodules were immersed in tissue freezing medium (Leica Byosistems, Germany) and frozen in liquid nitrogen. The 2 µm thick cryosections were made with cryotome (Leica Byosistems), placed onto glass slides and visualized under the light microscope. For alcian blue staining of SF scaffold cADMMSC, the medium was removed from the wells. Whole SF scaffolds were immersed in tissue freezing medium (Leica Byosistems) and frozen in liquid nitrogen. The 18 µm thick cryosections were made with a cryotome (Leica Byosistems) and placed onto glass slides coated with 1% poly-L-lysine (Sigma-Aldrich, Germany). Slides were fixed in 4% paraformaldehyde solution for 10 minutes at 4°C, rinsed once with distilled water (dH2O), incubated in alcian blue for 5 minutes at room temperature, rinsed again with dH2O and dried at room temperature. Slides were then mounted with histology mounting medium containing DAPI (Sigma-Aldrich), covered with coverslips and visualized under light and fluorescent microscope.
Light and fluorescent microscopy
For analysis of multilineage differentiation potential of cADMMSC and alcian blue staining of SF film and SF scaffold cADMMSC, an inverted microscope (Nicon Eclipse TS100, Nikon, Japan) and fluorescent microscope (Nicon Eclipse 80i, Nikon) equipped with Nikon Digital Sight DS-U2 camera were used. For analysis of DAPI staining, ultraviolet (UV) fluorescence filter of wavelength 330–380 nm was used. Images were captured in NIS–Elements D3.2 Live quality program.
Scanning electron microscopy
Scanning electron microscope (FEG-SEM, JEOL JSM 7600 F, Japan) under low voltage imaging conditions was used to display adhesion and morphology of cADMMSC seeded onto SF films and scaffolds. Images of SF film and SF scaffold cADMMSC were compared to the images of positive and negative control cADMMSC. Before the examination, the samples were fixed following the protocol described elsewhere.38,39 Briefly, the medium was removed from culture vessels. Wells were rinsed once with DPBS and fixed with 2.5% glutaraldehyde solution (Sigma) overnight. After fixation, the samples were rinsed with 0.1 M cacodylate buffer (Sigma) at pH 7.4 for 1 hour with three changes, rinsed with distilled water for 1 min and then dehydrated in 10 minute steps in a series of ascending ethanol baths (25%, 50%, 75%, 95%, and 100%). Dehydrated samples were then immersed into hexamethyldisilazane bath (5 minutes, 100% HMDS) (Sigma), air-dried, mounted, and sputtered with gold.
Scanning electron microscopy (SEM) was also used for the estimation of film thickness and for pore size evaluation in SF scaffolds. Film thickness was measured from the cross-section image of SF film. Pore size was estimated by measuring the pore diameter in two directions, parallel and perpendicular to the surface of the scaffold. Diameters of all pores that were not touching the edge of an image were measured by ImageJ software on three low magnification images, where approximately a third of the scaffold is visible.
RNA isolation
RNA was isolated from SF film cADMMSC, positive control cADMMSC, and negative control cADMMSC. SF film and positive control cADMMSC were detached from the SF film and polystyrene surfaces using cell scrapper. Negative control cADMMSC were detached by trypsinisation. The cell suspension was removed from the wells and centrifuged at 1600 r/min for 4 minutes. Pellet of cells was resuspended in DPBS and centrifuged again. Pellet of cells was then homogenized in 150 µL Trizol (ThermoFisher, USA) with a homogenizer (IKA T10 basic, Germany). Total RNA extraction was carried out using Trizol according to the manufacturer’s protocol. The amount of extracted total RNA was measured by UV spectrophotometer (ThermoFisher) at 260/280 nm wavelength.
Reverse transcription quantitative polymerase chain reaction
Two-step reverse transcription quantitative polymerase chain reaction (RT qPCR) for SF film cADMMSC, positive control cADMMSC, and negative control cADMMSC was performed. First, 1 µg of total RNA of each specimen was reverse transcribed into cDNA using High Capacity cDNA Reverse Transcription Kit with RNAse Inhibitor (ThermoFisher) according to the manufacturer’s protocol. Negative reverse transcription controls were included in each PCR run. All reactions were conducted in a total volume of 20 µL. Conditions for reverse transcription were as suggested in the manufacturer’s protocol: 25°C for 10 minutes, 37°C for 120 minutes, 85°C for 5 minutes. In the second step, relative quantification was performed using TaqMan Universal PCR Master Mix with UNG (ThermoFisher) and TaqMan gene expression assays Sox9, Col1A1, Col2A1, and Acan. TBP was used as a reference gene (Table 2; ThermoFisher). All qPCR amplifications were conducted in triplicates in a total volume of 20 µL. A 20 ng cDNA was used as a template. The amplification was carried out in 96-well plates with a Light Cycler 96 (Roche Life Science) using the following program: 50°C for 2 minutes, 95°C for 10 minutes, and 40 cycles at 95°C for 15 seconds, 60°C for 60 seconds.
Symbols, names, and assay identification numbers of genes of interest and their role in MSC chondrogenesis.

MSC, mesenchymal stem cells.
Statistical analyses
All statistical analyses were performed with NCSS software package (Kaysville, UT, USA).
Experiments with chondrogenic differentiation were repeated five times with cells from different dogs for negative and positive controls, and six times with cells from different dogs for cells growing on SF films. Three experiments with different cells were performed with SF scaffolds.
All RT qPCR experiments were run in triplicates. The efficiency corrected double delta Ct method was employed to normalize gene expression values. 42 The expression levels of Col1A1, Sox9, Col2A1, and Acan in positive control cADMMSC and SF film cADMMSC were compared to the expression levels of Col1A1, Sox9, Col2A1, and Acan in negative control cADMMSC and results were analyzed by Kruskal–Wallis non-parametric test. Statistical significance was determined with p < 0.05.
Results
Culturing and multilineage differentiation of negative control cADMMSC
Adipose tissue was successfully collected from all animals. Under the light microscopy, cells with fibroblast-like morphology were observed in all samples the day after plating. Using SEM, negative control cADMMSC appeared flat with spread morphology and wide extensions (Figure 1).
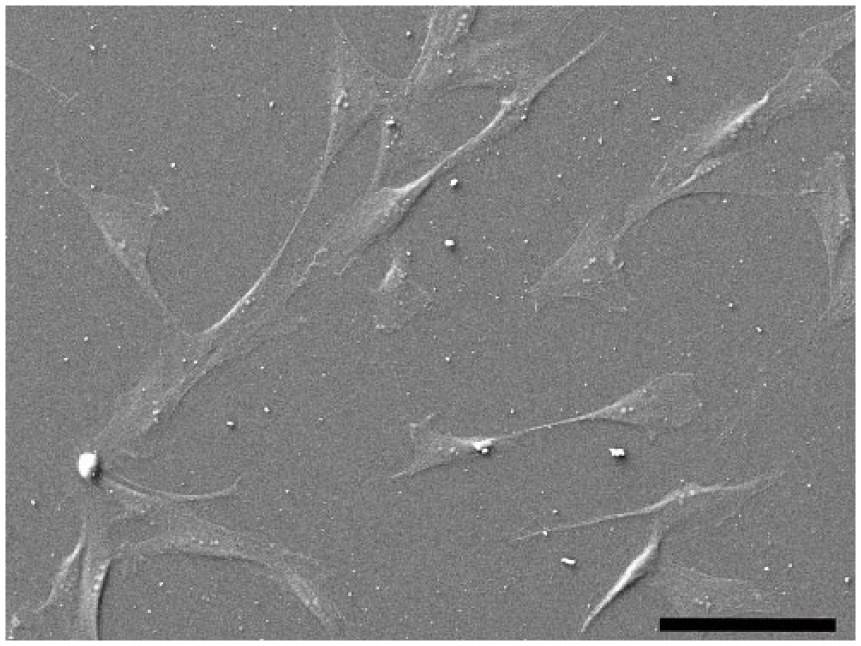
SEM image of negative control cADMMSC that appear flat with spread morphology and wide extensions.
Cell culture was maintained up to the second through the fourth passage. cADMMSC were capable of differentiating into adipocytes, osteocytes, and chondrocytes when cultured in specific differentiating media. After adipogenic differentiation, intracellular lipid droplet stained red using Oil-red-O. After osteogenic differentiation, mineral deposits in extracellular matrix stained red using Alizarin-red-S and after chondrogenic differentiation, proteoglycans in the extracellular matrix of layered cell clusters stained positive with alcian blue (Figure 2).
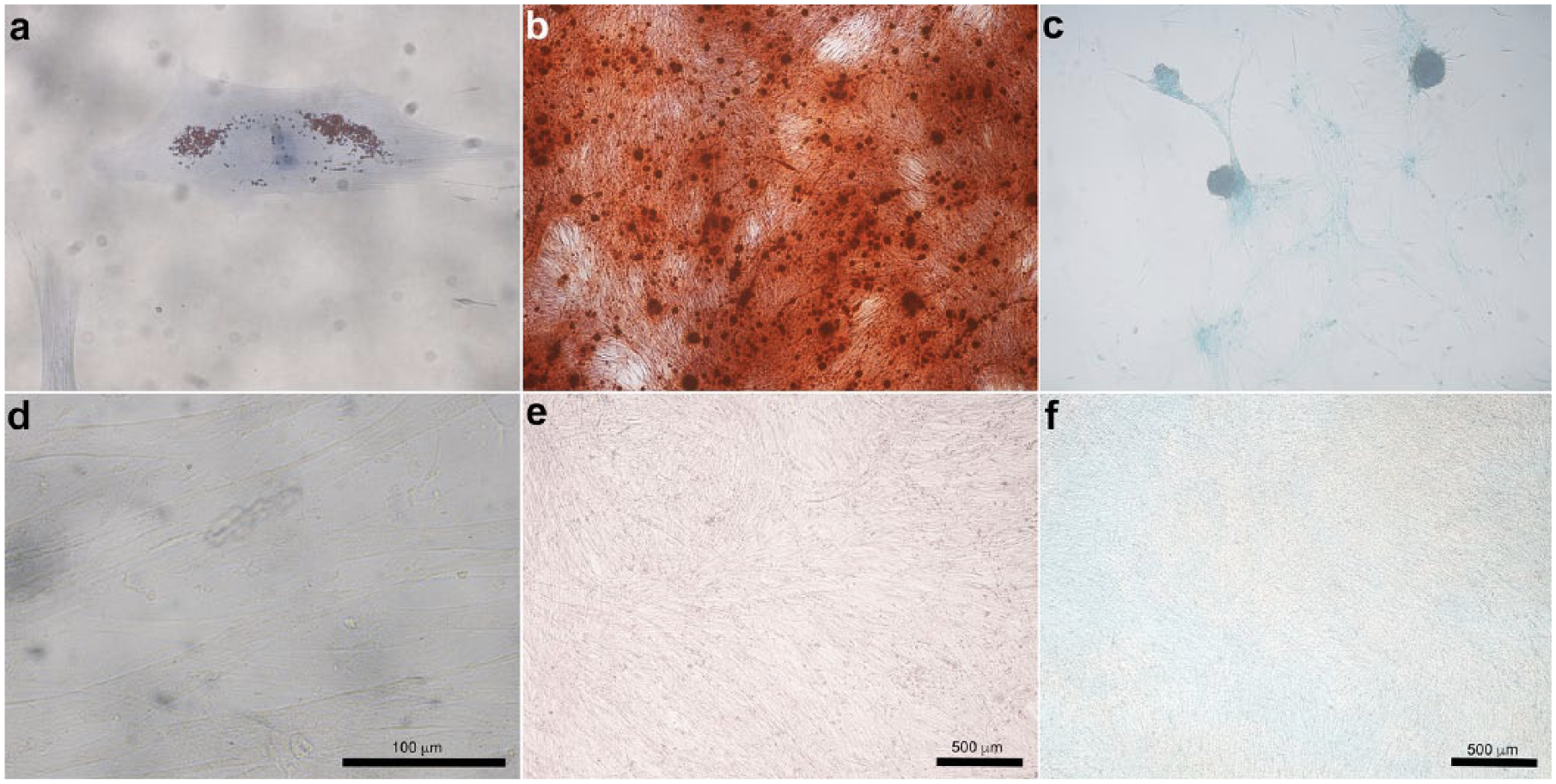
Differentiation potential of cADMMSC. Adipogenic potential of cADMMSC is indicated by red intracellular lipid droplets using Oil-red-O (a). In osteogenic differentiation, mineral deposits in extracellular matrix are stained red using Alizarin-red-S (b). Chondrogenic differentiation is indicated by the formation of chondrogenic nodules that stain blue with alcian blue (c). Respective negative controls are shown at the bottom (d, e, f).
Morphology and alcian blue analysis of positive SF film cADMMSC
SF film cADMMSC were cultured in normal culture medium on SF films. SF films prepared from SF solution with a concentration of 12.5 mg/mL were transparent, with diameter of 21 mm, and the approximate dry film thickness of 80 µm. The surface of the film appears smooth and dense, some roughness is visible only on a submicron level caused by nanosized pores that are assumed to form during drying of the film (Figure 3). The contact angle of a water droplet on a dry SF film is 70°, indicating a hydrophobic surface. After washing the film with PBS, the surface becomes hydrophilic with a water contact angle of 0°.

SEM images of SF film surface (a), the inset shows nanosized surface porosity at higher magnification, and cross-section of SF film (b).
SF film cADMMSC were cultured in normal cell culture media on SF films. SF films were transparent, with diameter of 21 mm, approximate thickness of 900 µm and average fibroin density of 12.5 mg/mL. Cells successfully adhered onto SF film surface. In comparison to negative control cADMMSC (Figure 1), SF film cADMMSC displayed less spindle-shaped morphology (Figure 4(a)). A tendency toward cell grouping was observed. The second day after seeding, nodule-like structures began to form (Figure 4(b)).
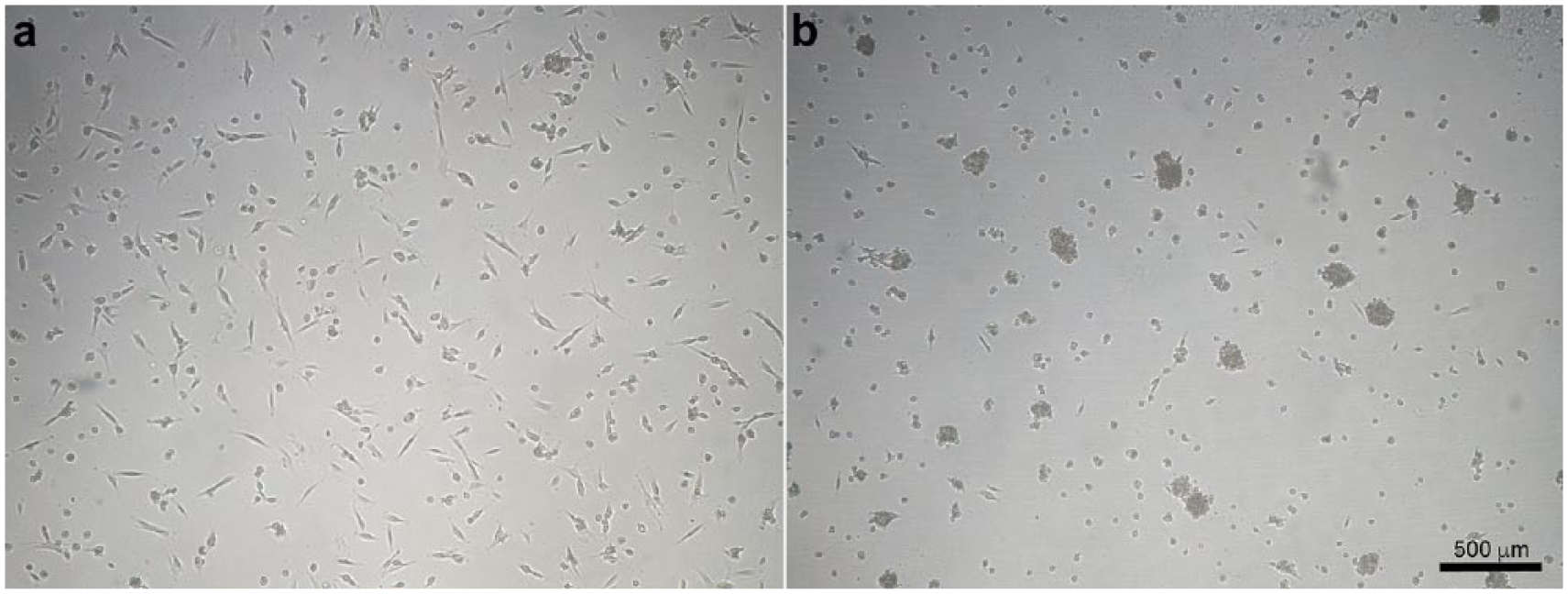
Morphology of SF film cADMMSC. The first day after seeding, SF film cADMMSC successfully attached to the SF film surface (a); Cells appeared less spindle-shaped compared to negative control cADMMSC. Many round-shaped cells are present and a tendency toward cell grouping was observed. On the second day after seeding, nodule-like structures began to form (b).
Positive control cADMMSC were cultured in the chondrogenic medium on a standard polystyrene surface for 14 days. Cells changed their morphology from adherent monolayer spindle-shaped cells to layered nodule-like cell clusters. Nodules that morphologically resembled chondrogenic nodules tended to form connections among each other and stained positive with alcian blue (Figure 5(a)). SEM images of positive control cADMMSC confirmed chondrogenic nodule–like structures with many extracellular matrix vesicles (Figure 5(d)).
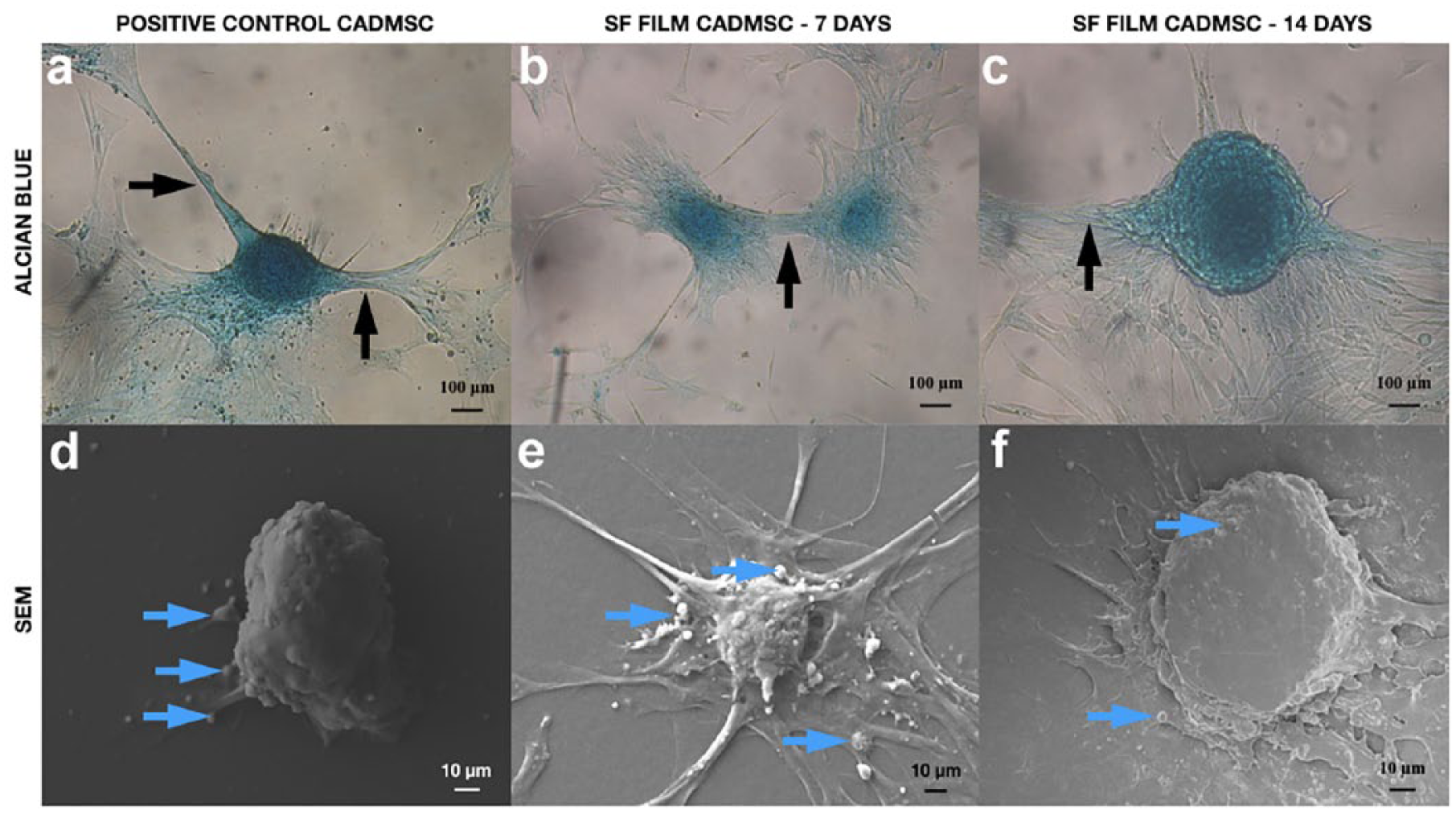
Morphology of positive control cADMMSC and SF film cADMMSC. In both, positive control cADMMSC (a, d) and SF film cADMMSC (b, e—7 days culture; c, f—14 days culture), clusters of cells reminding of a nodule were formed and stained blue with alcian blue. In both groups, interchondrogenic nodule connections were observed (black arrows). No apparent difference in shape or alcian blue staining intensity between presumed chondrogenic nodules in positive control cADMMSC (a) and SF film cADMMSC (b, c) were present. SEM analysis (d, e, f) confirmed the formation of chondrogenic-like nodules, which appeared more defined in positive control cADMMSC (d) and SF film cADMMSC cultured for 14 days (f) than in SF film cADMMSC cultured for 7 days (e). Many matrix vesicles are seen in positive control cADMMSC and SF film cADMMSC (blue arrows).
One to three days after culturing cADMMSC on SF films, chondrogenic-like nodules appeared (Figure 5(b) and (c)), similar to those formed in positive control cADMMSC (Figure 5(a)), with indistinguishable intensity of alcian blue staining. No apparent difference in alcian blue staining between SF film cADMMSC cultured for 7 and 14 days was detected (Figure 5(b) and (c)). SEM analysis of SF film cADMMSC revealed chondrogenic nodule–like structures (Figure 5(e) and (f)), comparable to those formed in positive control cADMMSC (5d). Many extracellular matrix vesicles were also observed. Nodule-like structures in SF film cADMMSC cultured for 14 days (Figure 5(f)) appeared to be more defined in comparison to those cultured for 7 days (Figure 5(e)).
Cryosections of alcian blue stained chondrogenic-like nodules from positive control cADMMSC revealed the correct round shape of a nodule, enclosed within a capsule-like structure. Nodule fibers appeared thick and homogenously arranged (Figure 6(a)). Cryosections of alcian blue stained chondrogenic-like nodules from SF film cADMMSC revealed more irregularly shaped nodules without an apparent capsule compared to cryosections of positive control chondrogenic nodule. Nodule fibers were thinner and organized into smaller separate circles formed inside of a nodule (Figure 6(b)).
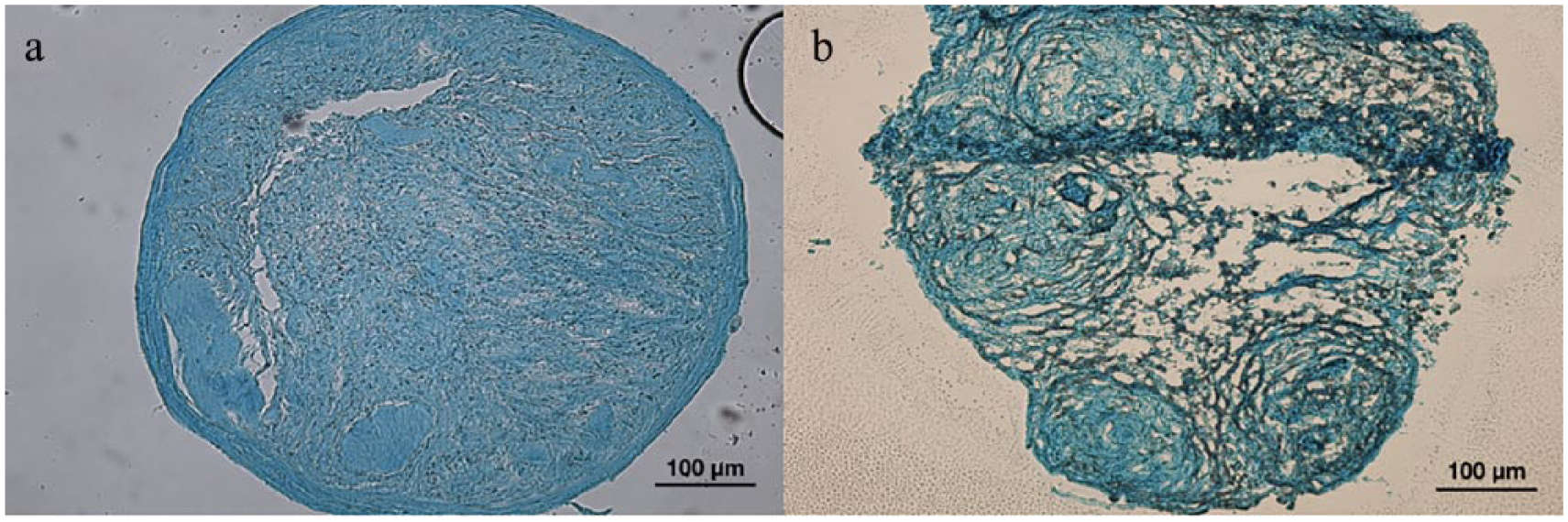
Morphology of cryosected chondrogenic nodules: (a) Cryosection of a chondrogenic nodule from positive control cADMMSC: chondrogenic nodule is roundly shaped surrounded with a capsule-like structure. Nodule fibers are thick and homogenously arranged; (b) Cryosection of a chondrogenic-like nodule from SF film cADMMSC: Nodule is more irregularly shaped without an apparent capsule. Nodule fibers are thin and arranged into individual smaller circles seen inside of a nodule.
Gene expression of chondrogenic markers
The mRNA expression of Col1A1 was similar in all four samples (Figure 7(a), N = 5 for positive and negative control, and 6 for SF films after 1 and 2 weeks). mRNA expression of Sox9 (Figure 7(c), N = 5 for positive and negative control, and 6 for SF films after 1 and 2 weeks) and Acan (Figure 7(d), N = 3 for positive control, 4 for negative control, and 6 for SF films after 1 and 2 weeks) was statistically significantly higher in positive control cADMMSC and SF film cADMMSC in comparison to negative control cADMMSC (p < 0.05 for Sox9 and p < 0.01 for Acan). The expression level of Col2A1 (N = 4 for positive and negative control, and 6 for SF films after 1 and 2 weeks) was not statistically significantly different between negative and positive control cADMMSC and SF film cADMMSC (Figure 7(b)).
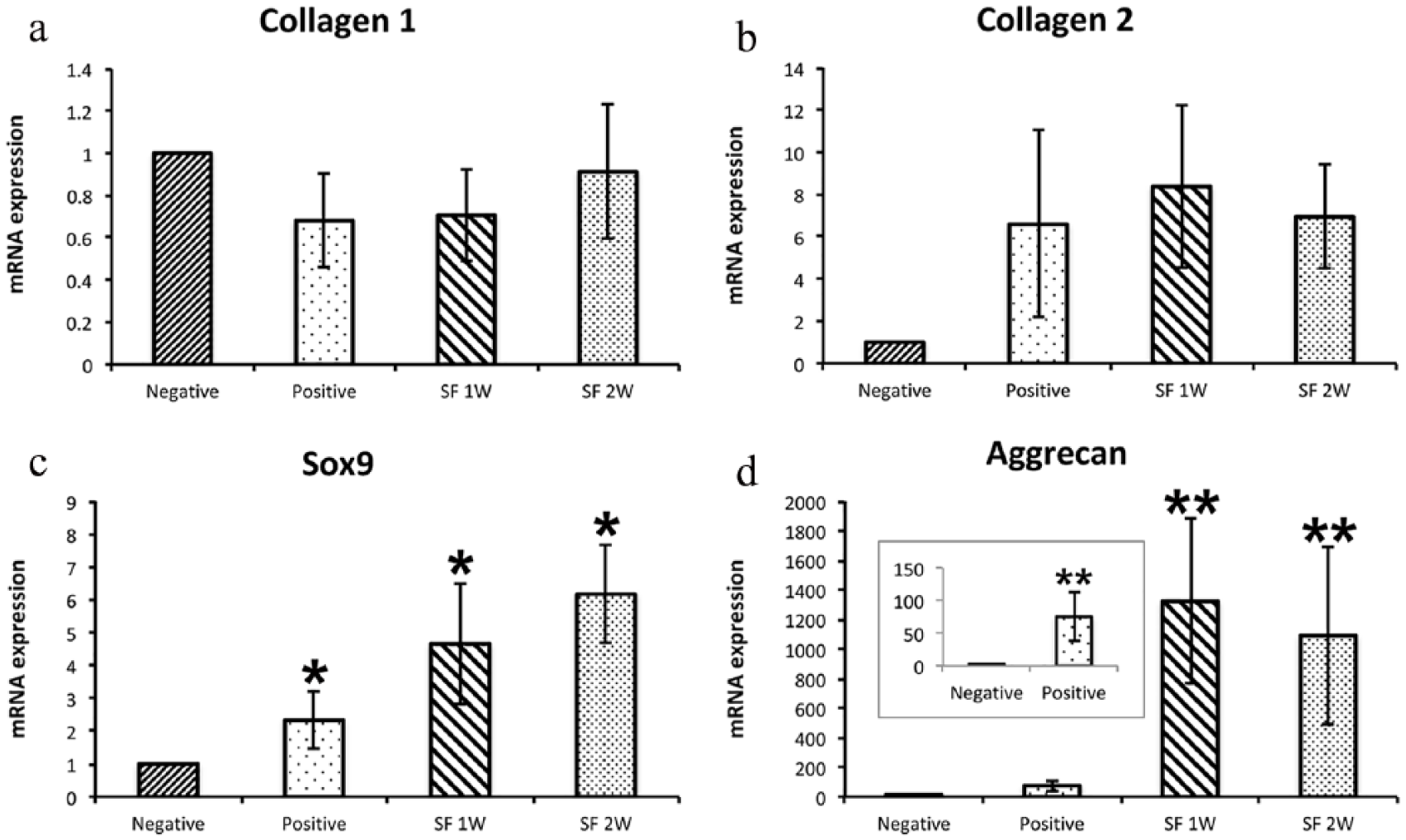
mRNA expression levels of Col1A1 (a) and ColL2A1 (b) were similar in all four groups (negative controls and positive controls (N = 5 for Col1A1 and N = 4 for Col2A1), and cells cultured for 1 week (SF 1W) or 2 weeks (SF 2W; N = 6 for all samples) on SF films. Expression levels of Sox9 (c, N = 5 for positive and negative controls and N = 6 for SF 1W and SF 2W) and Aggrecan (d, N = 3 for positive controls, N = 4 for negative controls and N = 6 for SF 1W and SF 2W) differed significantly between negative control and positive control groups, as well as between negative control and cells cultured for 1 or 2 weeks on SF films (*p < 0.05 for Sox9 and **p < 0.01 for Aggrecan). Results are presented as mean ± SEM and data were normalized to the expression of genes in negative control samples.
SF scaffold characteristics
The volume of the scaffolds was 0.3 mL with an approximate thickness of SF scaffolds 5.2 mm. Light microscopy images of unseeded cryosections of scaffold showed a porous matrix with interconnected elongated pores (Figure 8(a) and (b)). Scaffold porosity was 92.7%, of which 84.6% represents open, and 8.1% closed porosity. The mean pore opening diameter measured as a function of pressure from the mercury intrusion measurements was 50 µm, with a minimum and maximum at 0.004 and 107 µm, respectively. SEM images of nonseeded SF scaffold revealed a porous matrix with a broad distribution of pore sizes, where the bottom side of a scaffold had larger pores (Figure 8(c)). Mean pore diameter measured on SEM images in the elongated direction was 314 ± 296 µm and 58 ± 55 µm in the direction parallel to the surface.

Morphology of a non-seeded SF scaffold: (a) Light microscopy image of a cryosection of an upper part of the scaffold with a pore size of about 100 µm; (b) light microscopy image of a cryosection of a bottom part of the scaffold with a pore size up to 455 µm; and (c) SEM image of the scaffold. The green arrow indicates the upper part of the scaffold. Orange arrow indicates the bottom part of the scaffold.
Morphology and alcian blue analysis of positive, SF scaffold cADMMSC
SF scaffold cADMMSC were cultured in culture medium in SF scaffolds. Cell migration into the scaffold was analyzed by a fluorescent microscope with DAPI staining (Figure 9(c) and (d)) and SEM (Figure 10). Based on the presence of the cells in all cryosections, it was concluded that SF scaffold structure allows penetration of cells inside the scaffold, although more cells remained on the surface of the scaffold (Figure 9). Nevertheless, some cells migrated throughout the scaffold and successfully attached to the pore walls. Positive alcian blue staining of cells was confirmed on the surface and within the scaffold (Figure 9(a) and (b)). Chondrogenic nodules were observed only occasionally close to the surface of the scaffold (Figure 9(b)). Mostly, amorphous layers of cells were observed, but these also stained positive with alcian blue (Figure 9(a)).
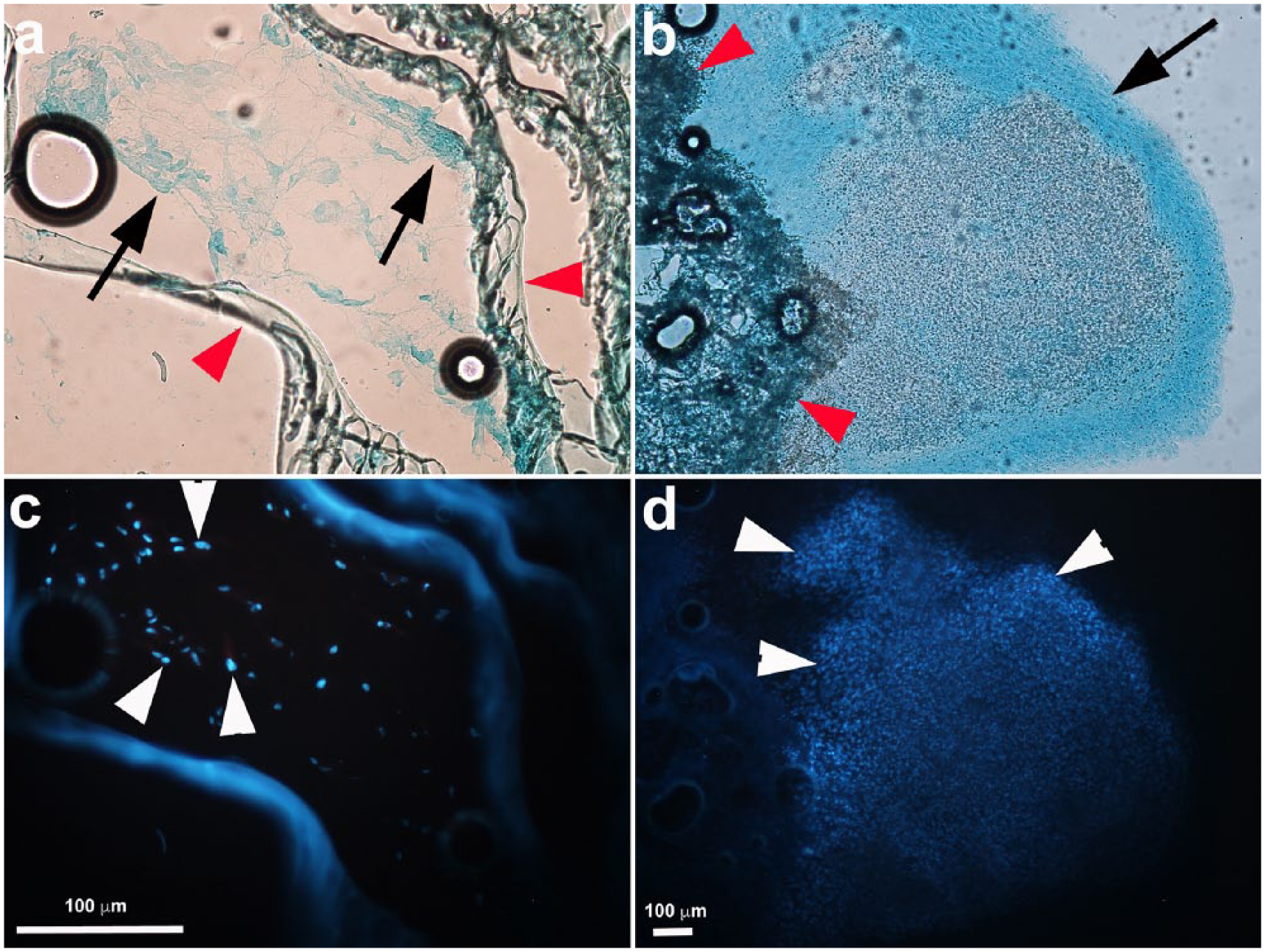
SF scaffold cADMMSC. In upper row, alcian blue staining of scaffold cryosections is shown. In lower rows, the same images are shown under fluorescence, where cell nuclei were stained with DAPI (white arrowheads). In parts of the scaffold surface, chondrogenic nodule–like structures (black arrow) were present (b) and cell density in these structures was very dense (white arrowheads—cell nuclei stained with DAPI in panel (d)). Inside the scaffold (panels a and c) cells were present, but at lower density in comparison to surface (white arrows—cell nuclei stained with DAPI in (c) and (d)) and they did not form chondrogenic nodules, although they stained positive with alcian blue (arrow, a). SF structure is marked with red arrowheads in panels a and b.

SEM images of cADMMSC grown on SF scaffold: (a) cells attached to the pore wall. Numerous matrix vesicles and fibers are seen on top of the cells; (b) cell inside the scaffold pore at higher magnification. Matrix vesicles are protruding from the cell; (c, d) Same cells marked with color.
SEM analysis of SF scaffold cADMMSC confirmed homogeneous distribution of cells throughout the scaffold and their attachment to the pore walls. A dense network of fibers was formed, and numerous extracellular matrix vesicles were observed (Figure 10(a) and (b)—scaffold with cells at different magnifications).
Discussion
In attempts to mimic the native extracellular matrix, numerous studies are focusing on culturing MMSC in a 3D culture system, which better imitates their natural environment. Although several biomaterials have been shown to promote differentiation of MMSC toward different lineages,21–25 there are no reports about SF inducing differentiation of MMSC. In the present study, we demonstrate that cADMMSC presumably undergo chondrogenic differentiation when grown on SF films and SF scaffolds in a standard cell culture medium. cADMMSC appeared to follow chondrogenesis without chondrogenic-specific conditions such as high cell density or stimulation with TGF-β. 4 Morphology of cells observed under the microscope, positive alcian blue staining of the SF film cADMMSC and SF scaffold cADMMSC, and upregulation of Sox9 and Aggrecan mRNA expression in cADMMSC on SF films suggest that cells underwent chondrogenic differentiation. Chondrogenesis of cADMMSC cultured on SF films was also suggested by similar chondrogenic-like nodule morphology, and a tendency to form interchondrogenic nodule connections as it was observed in positive control cADMMSC. In comparison to cells cultured on SF films, cADMMSC cultured inside the SF scaffolds formed chondrogenic-like nodules only in parts of the scaffold. Elsewhere, amorphous layers of cells were present, but, interestingly, both nodules and amorphous layers of cells inside the SF scaffolds stained positive with alcian blue suggesting at least partial induction of chondrogenesis. Comparison of SEM images between positive control cADMMSC, SF film cADMMSC, and SF scaffold cADMMSC showed the presence of cell nodules that resembled chondrogenic nodules. In all three groups of cells, many extracellular matrix vesicles were present and abundant extracellular matrix formed which appeared as chondrogenic-like nodule structures. SEM images of SF scaffold cADMMSC also revealed numerous fibers formed by the cells, which suggest possible formation of collagen.
It has been previously demonstrated that in vitro initial cell aggregates, cultured on polystyrene surface under specific culture conditions, contain Col1A1. Within 2–3 weeks of chondrogenic differentiation, MMSC undergo chondrogenic differentiation and start to produce abundant extracellular matrix composed of Col2A1. 5 Differentiation of MMSC is characterized by a decrease in proliferation and upregulation of lineage-specific genes. 43 TGF-β is known to induce chondrogenesis by activating SMAD signaling pathway and upregulating chondrogenic genes such as Sox9. 1 The latter has been identified as the main transcriptional regulator of chondrogenic specific markers, namely, type II collagen. 40 Another marker of chondrogenic differentiation is Aggrecan, an essential component of mature cartilage, whose expression also increases during chondrogenic differentiation of MMSC. 44 In our study, the expression of Col1A1 and cartilage-specific markers Sox9, Aggrecan, and Col2A1 were determined by RT qPCR in cells grown on SF films and positive and negative controls. mRNA expression of Col1A1 and Col2A1 was not significantly different between positive and negative controls cADMMSC and SF film cADMMSC. However, both Aggrecan and Sox9 mRNA expression was statistically significantly upregulated in positive control cADMMSC and SF film cADMMSC. This suggests that chondrogenesis was indeed initiated also at the molecular level. Currently, it is difficult to speculate why we did not also detect increased expression of Col2A1. However, it has to be noted that mRNA expression of Col2A1 was very low and perhaps, the time of chondrogenesis was too short to induce also Col2A1, a marker of mature chondrocytes. The RT qPCR results of our study thus indicate that chondrogenic differentiation of SF film cADMMSC is comparable to positive control cADMMSC. Based on the upregulation of Sox9 and Aggrecan in both positive control and SF film cADMMSC, we concluded that the initial stages of chondrogenesis took place, but it might take longer than 14 days for Col2A1 to be significantly upregulated. Due to protein overload and lower number of cells when isolating RNA from cells with proteins from the scaffold, we were unable to purify enough RNA perform RT qPCR from SF scaffold cADMMSC to confirm mRNA expression results also with cells grown in SF scaffolds.
Mechanisms behind presumed chondrogenic differentiation of cADMMSC on SF in our study are not yet known. In line with the high cell density as one of the conditions needed for MMSC differentiation, the study of Dudakovic et al. 8 showed that maintaining adipose-derived human MMSC in confluent cultures leads to post proliferative conveyance of more specialized cellular phenotypes expressing osteogenic, chondrogenic, and adipogenic biomarkers. Similarly, Bosnakovski et al. 6 showed that bovine bone marrow–derived MMSC cultured in a pellet culture system also differentiates into chondrocytic lineage. In the present study, the number of cells seeded on SF films was only 1 × 104 and did not reach confluency. Since cells started to differentiate the day after their attachment on SF films, high cell density was excluded as a possible reason for presumed chondrogenic differentiation of MMSC on SF films. High cell density, however, might have contributed to the chondrogenesis of SF scaffold cADMMSC, since cell seeding density was higher (9 × 5 µL droplets of 5 × 105 cells per scaffold) due to the higher overall surface for cell attachment. Besides high cell density, culture medium could also play a role in chondrogenic MMSC differentiation even without specific factors added to the culture media. Fortier et al. 45 reported sporadic chondrogenesis of equine MMSC cultured with culture medium supplemented with 10% FBS. Since chondrogenesis did not occur in a serum-free medium in this study, the reason for chondrogenic differentiation was attributed to bioactive factors present in the FBS. In our study, FBS is unlikely to be the reason for chondrogenic differentiation of MMSC, as the chondrogenic differentiation of negative control cADMMSC, cultured on a standard polystyrene surface in a cell culture medium with FBS, was never observed.
Mechanical properties of SF are also unlikely the reason for presumed chondrogenic differentiation of cADMMSC since chondrogenesis seemed to occur not only on 2D SF films but also on architecturally different 3D SF scaffolds. Passage number was shown before to affect the differentiating behavior of cells. In human donors, spontaneous chondrogenesis was reported in early passages of human periosteum-derived cells, which are otherwise known to have bilineage potential. 7 In our study, cells from second to fourth passage were always used and therefore, an early passage might have contributed to the differentiation of cADMMSC grown on SF films. However, this was clearly not the main reason for presumed chondrogenic differentiation of cells grown on SF films as negative control cADMMSC cells did not differentiate into chondrocytes, despite being from the same passages. In addition, source of MMSC might also contribute to their spontaneous phenotypic changes. Naruse et al. 9 showed that rat cells derived from fetal circulating blood spontaneously differentiated into both chondrocytic and osteocytic cells on plastic culture dishes in a standard culture medium. However, rat bone marrow–derived MMSC did not undergo similar phenotypic changes under the same conditions. In our study, cADMMSC were used, which are rarely studied. There are no reports about spontaneous differentiation of cADMMSC. Therefore, it is not possible to speculate whether the tissue source of MMSC in our study contributed to the presumed chondrogenic differentiation of cADMMSC cultured on SF films without specific growth factors present. Surely, this cannot be the main reason as we never observed chondrogenic differentiation in negative controls even though the cells were obtained from the same donors. One possible explanation for presumably chondrogenic induction in our experiment would be hydrophobicity of SF films. Indeed, SF films are initially very hydrophobic, but when wetted, they form a hydrophilic hydrogel with a water contact angle of 0°. Therefore, when cells were seeded on SF films, the surface was hydrophilic and is unlikely to force cells into cell–cell adhesion. However, the surface of SF films has not been biochemically characterized and we do not know anything about cell adhesion processes on SF films. Therefore, in the future studies, it would be interesting to study cell adhesion molecules (such as integrins) and processes to determine how cells adhere to the SF and whether this contributes to the differentiation of cells on this biomaterial.
To find out whether there are differences between donor species regarding MMSC differentiation on SF films, a corresponding study of ADMMSC from different animal species should be conducted in the future. It is difficult to speculate about the reasons for cADMMSC differentiation in the absence of specific growth factors as the literature on this topic is sparse. Only a handful of studies reported a chondrogenic differentiation of MMSC either on a standard plastic surface or on biomaterials in the absence of special growth factors and/or high cell density seeding. Different parameters used in each study such as donor species, tissue source of MMSC, culture media composition, and culturing methods makes difficult any direct comparisons of these studies. Future studies are therefore necessary to determine which factors might contribute to the mechanisms lying behind chondrogenic differentiation of MMSC in the absence of specific growth factors.
Results of our study show that SF could be considered as a promising biomaterial not only for culturing cells but also for the induction of controlled chondrogenesis of MMSC. None of the previous studies using SF has shown its ability to guide MMSC toward chondrogenic differentiation, so this might be a species-specific effect, as our study was the first study to examine the growth of canine MMSC on SF. This effect of SF on MMSC in our study, therefore, represents a basis for further studies aiming to understand the key factors and mechanisms responsible for induction of differentiation of MMSC into chondrocytes.
Footnotes
Data availability
The raw data and processed data required to reproduce these findings are available from the authors upon request.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship and/or publication of this article: This study was supported by grant P4-0053 from Slovenian research agency and Metka Voga is supported by a graduate fellowship from the Slovenian research agency.