
Abstract
Mesenchymal stem cells (MSCs) are a potential source of cell-based disease-modifying therapy in Parkinsonian disorders. A promising approach to develop in vitro culture methods that mimic natural MSC niche is cell priming. Uric acid (UA), a powerful antioxidant, scavenges reactive oxygen species, which has a vital role in maintaining self-renewal and differentiation potential of MSCs. Here, we demonstrated that UA treatment in naïve MSCs stimulated glycolysis and upregulated transcriptional factors responsible for regulation of stemness, leading to increase in the expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes. UA-primed MSCs had more enhanced neuroprotective properties in cellular and parkinsonian animal models compared to naïve MSCs by inhibiting apoptotic signaling pathways. Additionally, expression of miR-137 and miR-145 was decreased in UA-treated MSCs. Our data demonstrated that priming MSCs with UA augment neuroprotective properties through enhanced self-renewal and differentiation potential, suggesting a practical strategy for improving the application of MSCs in parkinsonian disorders.
Introduction
Parkinson’s disease (PD) is characterized pathologically by the progressive loss of dopaminergic neurons in the substantia nigra (SN) and the presence of Lewy bodies, proteinaceous fibrillar cytoplasmic inclusions that are mainly composed of aggregated α-synuclein.1,2 The mainstay of PD management is symptomatic treatment with drugs that increase dopamine concentrations or directly stimulate dopamine receptors. 3 Nevertheless, these therapies do not affect the progressive nature of PD and, moreover, they are ineffective against some axial parkinsonian symptoms and various non-motor symptoms. Thus, it is crucial to develop disease-modifying treatments that reduce the rate of neurodegeneration or stop the disease process in PD. Recently, the concept of stem cell therapy has been extended to adult stem cells, which secrete biologically active molecules, exerting beneficial effects on their surroundings.4–6 Previous studies have shown that mesenchymal stem cells (MSCs) can act as potent modulators of PD-related neurodegenerative microenvironments through the modulation of neuroinflammation, inhibition of apoptosis, increased neurogenesis and neuronal differentiation, enhancement of autophagy, and modulation of α-synuclein propagation.7–10 However, the major challenge in MSC-based therapies is to develop in vitro culture systems that mimic the natural MSC niche, while allowing clinical scale cell expansion without compromising quality and function of cell.11–13 To date, several studies have demonstrated that the modulation of biological, biochemical, and/or biophysical factors can influence the fate, lineage-specific differentiation, functions, and therapeutic potential of MSCs.12,14,15 One approach is cell priming. Many studies have demonstrated the effects of MSC priming with hypoxia, cytokines, growth factors, pharmacological or other chemical agents, biomaterials, and different culture conditions.5,11,16
Uric acid (UA), a purine metabolite, is a powerful antioxidant, which can be found intracellularly and in all body fluids.17–19 It not only scavenges reactive oxygen species (ROS) but also blocks the reaction of the superoxide anion with nitric oxide, which can injure cells by nitrosylating the tyrosine residues of proteins. It also prevents extracellular superoxide dismutase degradation. 20 Ample evidence has suggested that UA has neuroprotective properties in PD, showing that PD patients with higher UA levels have been linked to reduced risk of PD incidence as well as slower disease progression.19,21,22 The beneficial effects of UA have also been observed in other neurodegenerative diseases, such as amyotrophic lateral sclerosis, 23 Alzheimer’s disease, 24 and Huntington’s disease. 25 Hence, UA has not only antioxidant properties but also neuroprotective properties against neurodegenerative conditions.
Stemness encompasses the maintenance of self-renewal and multi-lineage differentiation potential.26–28 The functions fulfilled by specialized stem cells, such as stem cell proliferation, lineage specification, and quiescence require a certain energy supply.29,30 Glycolysis is the enzymatic conversion of glucose to pyruvate, which generates two net ATP molecules per glucose molecule. 31 However, cells in oxygen-rich environments may prefer oxidative phosphorylation (OXPHOS), which leads to a more efficient ATP production by oxidizing pyruvate to acetyl-CoA in the mitochondrial tricarboxylic acid cycle.31,32 Many types of stem cells rely on glycolysis when they are undifferentiated, but they activate the mitochondrial OXPHOS process during differentiation.33,34 Stimulation of glycolysis in pluripotent or adult stem cells by hypoxia or supplementation with insulin promotes stemness, while glycolysis inhibition halts proliferation and induces cell death. 35 In terms of pluripotency genes, OCT4, NANOG, and SOX2 constitute the core regulatory network that suppresses differentiation-associated genes, thereby maintaining cell pluripotency.36,37 OCT4, a key transcription factor essential for self-renewal and survival of MSC interacts with other embryonic regulators, such as SOX2 and NANOG, to regulate the network that maintains pluripotency and inhibits differentiation. 38 Moreover, OCT4 has a number of targets associated with energy metabolism, which may impact the balance between glycolysis and oxidative metabolism. 39
ROS resulting from cellular metabolism are crucial for stemness and stem cell differentiation. Differentiation stimuli cause elevated ROS levels, thus inducing stem cell differentiation into specific lineages.40,41 However, glycolysis enhancement via hypoxia and OXPHOS suppression, which lead to concomitantly decreased ROS levels, promote stem cell maintenance and proliferation, thereby repressing differentiation. 42 On the other hand, ROS can also lead to stem cell dysfunction, leading to senescence with loss of stemness.43,44 Thus, antioxidant would be a core relationship between redox homeostasis and pluripotency of stem cells. In the present study, we hypothesized that UA can efficiently decrease ROS levels against oxidative stress and thus play a central role in stemness maintenance. To prove this, we evaluated whether UA treatment enhances stemness properties in MSCs. Moreover, we tested whether UA-primed MSCs exerts a neuroprotective effect in PD animal models, thus providing a strategy to improve MSC application in tissue engineering.
Materials and methods
MSC and SH-SY5Y cultures
Frozen vials of characterized human MSCs at passage two were obtained from the Severance Hospital Cell Therapy Center (Seoul, South Korea; IRB: 4–2008-0643). MSCs were maintained in low-glucose Dulbecco’s Modified Eagle Medium (DMEM, HyClone), supplemented with 10% fetal bovine serum (FBS; HyClone) and an 1% penicillin and streptomycin (P/S; HyClone). A human neuroblastoma cell line possessing characteristics of dopaminergic neurons, SH-SY5Y, was obtained from the Korean Cell Line Bank (Seoul, South Korea). 45 SH-SY5Y cells were maintained in high-glucose DMEM, supplemented with 10% FBS and an 1% P/S. Both MSCs and SH-SY5Y cells were cultivated in a humidified incubator at 37°C and 5% CO2 before use. For priming MSCs, the cells were plated at a density of 1 × 104 per square centimeter 46 and treated with either 200 μM or 400 μM UA (Sigma), each for 24 h and 48 h. The SH-SY5Y cells were plated at a density of 2 × 104 per square centimeter 46 and treated with 100 µM 1-methyl-4-phenylpyridinium ion (MPP+; Sigma) for 24 h. Subsequently, the cells were cocultured with MSCs or priming MSCs for 24 h.
Mice
All procedures were performed in accordance with the Laboratory Animals Welfare Act, the Guide for the Care and Use of Laboratory Animals, and the Guidelines and Policies for Rodent Experimentation provided by the Institutional Animal Care and Use Committee (IACUC) at the Yonsei University Health System. Male C57BL/6 mice (Orient Bio) were acclimated in a climate-controlled room with a constant 12/12-h light/dark cycle for 1 week prior to drug administration.
MTS assay
Cell viability was measured using the CellTiter 96® AQueous One Solution Cell Proliferation Assay (Promega), in accordance with the manufacturer’s protocol. The absorbance was measured using a SpectraMax 340PC384 microplate reader (Molecular Devices) at 490 nm, and the data were analyzed using the Softmax Pro software (Molecular Devices).
MSC differentiation and immunostaining
Osteogenic differentiation was initiated by plating MSC at a density of 5 × 103 cells per square centimeter and induced by adding ascorbic acid (50 μg/ml; Sigma), sodium β-glycerophosphate (10 mM; Sigma), and dexamethasone (10−8 M; Sigma) to the complete medium. After 2 weeks, the plates were washed with phosphate buffered saline (PBS, HyClone), fixed with 4% paraformaldehyde, and stained with 1% Alizarin Red S (ARS; Sigma). Adipogenic differentiation was initiated by plating MSC at a density of 5 × 103 cells per square centimeter and induced by addition of dexamethasone (10−7 M; Sigma) and insulin (6 ng/ml; Gibco) to the complete medium. After 3 weeks, the plates were washed with PBS, fixed with 4% paraformaldehyde, and stained with Oil red O (ORO; Sigma). Chondrogenic differentiation was initiated by plating MSC at a density of 5 × 103 cells per square centimeter and induced by ascorbic acid (50 μg/ml; Sigma) and TGFβ-3 (1 ng/ml; R&D system) addition to the complete medium. After 2 weeks, the plates were washed with PBS, fixed with 4% paraformaldehyde, and stained with 0.05% Alcian Blue (AB; Sigma).
Quantitative real-time polymerase chain reaction (RT-PCR)
Total RNA was extracted from the MSCs using TRIzol reagent (Invitrogen), in accordance with the manufacturer’s protocol. An equal amount of RNA (1 µg), for each experiment, was reverse transcribed using High capacity cDNA reverse transcription kits (Thermo fisher). Subsequently, cDNA were used as a template for RT-PCR with the AccuPower® 2X GreenStar™ qPCR MasterMix (Bioneer). For miRNA quantification, reverse transcription was performed using the Mir-X miRNA First-Strand Synthesis Kit (Clontech Laboratories) and cDNA were used as a template for RT-PCR with Mir-X miRNA qRT-PCR SYBR Kit (Clontech Laboratories). Gene-specific primer sequences are listed in the supplemental tables (Supplemental Tables 1 and 2). All the RT-PCR was performed with the RT-PCR StepOnePlus system (Applied Biosystems). Data were normalized to GAPDH or U6 and analyzed by the 2–ΔΔCT method.
Western blotting
Cells and brain tissues were dissolved in ice-cold RIPA buffer plus a protease inhibitor cocktail (Sigma). The lysates were centrifuged at 4°C for 20 min (14,000 × g) and the supernatants transferred to fresh tubes. Briefly, 10 μg of protein were separated by SDS-gel electrophoresis and transferred onto hydrophobic PVDF membranes (GE Healthcare). The membranes were blocked in skim milk and then incubated overnight at 4°C with the indicated primary antibody. The primary antibodies are listed in the supplemental table (Supplemental Table 3). The membranes were washed in PBST followed by incubation for 2 h at room temperature (RT) with fluorescently conjugated goat anti-mouse or rabbit antibodies. The blots were visualized with an ImageQuant LAS 4000 min (GE Healthcare). Quantitation of western blots was performed using ImageJ.
ROS detection and immunostaining
The intracellular ROS levels were measured through the 2′,7′-dichlorodihydrofluorescein diacetate (DCFDA) cellular ROS detection assay kit (Abcam), according to the manufacturer’s instructions. The generation of fluorescent oxidized DCF was determined using a SpectraMax 340PC384 microplate reader at 520 nm, and the data were analyzed using the Softmax Pro software. For immunostaining, the cells were washed twice with PBS and fixed in 10% methanol in PBS for 30 min. The cell nuclei were counterstained with 4′,6-diamidino-2-phenylindole (DAPI; Invitrogen). The immunostained cells were analyzed using a Zeiss LSM 780 confocal microscope system (Zeiss).
L-lactate assay
The intracellular lactate levels were measured using a colorimetric L-lactate assay kit (Abcam), according to the manufacturer’s instructions. The absorbance was measured using a SpectraMax 340PC384 microplate reader at 450 nm, and the data were analyzed using the Softmax Pro software.
LDH cytotoxicity assay
LDH release was measured using the LDH cytotoxicity detection kit (Takara Bio, Cat. # MK401), according to the manufacturer’s instructions. The supernatant (100 µL) and reaction mix (100 µL) were mixed in 96-well plates and incubated for 30 min at RT. All samples were measured using a SpectraMax 340PC384 microplate reader at 490 nm, and the data were analyzed using the Softmax Pro software.
UA-enhancement therapy
To induce the hyperuricemia model, the mice were injected with potassium oxonate (PO; Sigma; intraperitoneal injection [i.p.], 250 mg/kg, daily) or inosine 5′-monophosphate disodium salt hydrate (IMP; Sigma; i.p. 500 mg/kg, daily). The mice were randomly divided into three groups (n = 12 per group); (a) control, (b) PO, (c) PO, and IMP. All the mice were sacrificed after 4 weeks.
Serum urate measurement
Blood samples were collected into EDTA tubes from the vena cava using a syringe. The blood samples were centrifuged at 4000 × g at 4°C for 20 min to obtain the serum as a supernatant. Serum samples (approximately 20 µl) were collected from each blood sample. The quantity of serum urate in 3.3 µl of each serum sample was measured using the Amplex Red Uric acid/Uricase assay kit (Molecular Probes), according to the manufacturer’s protocol.
Isolation and culture of mouse BM-MSCs
The femurs were dissected by cutting at the joints, carefully scrubbed to remove the residual soft tissues, and collected in a culture dish, containing the DMEM medium supplemented with 10% FBS and 1% P/S on ice. The bones are washed twice with PBS to flush away the blood cells and the residual soft tissues. A 27-gauge needle attached to a 1 mL syringe is used to draw 1 mL complete DMEM medium supplemented with 10% FBS and 1% P/S then the needle is inserted into the bone cavity. The marrow out is slowly flushed and the bone cavities washed twice again until the bones become pale. All the bone pieces are removed from the dish using forceps, leaving the solid mass in the medium, and the dish is incubated at 37°C in a 5% CO2 incubator for 5 days. 47
Animal experiments
To evaluate the modulatory effects of priming MSCs in a PD animal model, the mice were injected with 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP, Sigma; i.p., 30 mg/kg, daily for 14 days) that is well known to damage the nigrostriatal dopaminergic pathway as seen in PD. They were then randomly divided into five groups (n = 5 per group); (a) control, (b) MPTP, (c) MPTP and mouse control MSCs, (d) MPTP and mouse MSCs primed with PO + IMP, and (e) MPTP and human MSCs primed with UA. The mice in the MSC groups were injected with MSCs or primed MSCs via the tail vain (1 × 106 cells per 200 μl) at 3 days after the final MPTP injection. All the mice were sacrificed 4 weeks after MSC injection.
Brain sample preparation
All the mice were deeply anesthetized with chloral hydrate (Fluka; i.p., 0.4 g/kg) and perfused with 4% paraformaldehyde in 0.1 M phosphate buffer. The brains were embedded in paraffin, and 20 µm-thick coronal sections were cut and placed on slides.
Fluorescence activated cell sorting (FACS)
The human and mouse MSCs were characterized by FACS. Briefly, the cell suspensions were washed twice with PBS containing 0.1% BSA. 2 × 106 cells/ml were incubated with primary antibody at 4°C for 1 h and washed twice with PBS containing 0.1% BSA. The primary antibodies are listed in the supplemental table (Supplemental Table 3). The cells were analyzed by cytometric analysis using an LSR II flow cytometer (Beckman coulter) with BD FACSDiva software.
Immunohistochemistry
Deparaffinized brain sections were washed in PBS and incubated in 0.2% Triton X-100 for 15 min at RT. They were then blocked with 0.5% BSA for 30 min. After blocking, they were rinsed three times in 0.25% BSA and incubated overnight at 4°C with a primary mouse anti-tyrosine hydroxylase (TH) antibody (Sigma). The TH antibodies were detected via 0.05% diaminobenzidine (DAB, Vector Laboratories). The immunostained cells were analyzed by brightfield microscopy (Olympus).
Rotarod test
To assess motor function, coordination, and balance, the mice were tested on the Rotarod apparatus (MED-Associates). The day before the training session, the mice were habituated to the apparatus for 15 min. During the training trials, the mice were trained to run on the rotarod at 20 rpm for 10 min without falling, twice a day for three consecutive days. In the test trials, the mice were placed on the rotarod at 30 rpm and placed on the rotarod with increasing speed, from 5 to 40 rpm (maximum cut-off time: 700 s). The latency time to fall was recorded.
Stereological cell counts
The TH-stained dopaminergic neurons were counted in the SN of every fourth section throughout the SN pars compacta. The number of TH-stained cells was then counted at high power, starting at a random point. To avoid double counting of neurons with unusual shapes, the TH-stained cells were counted only when their nuclei were optimally visualized, which occurred only in one focal plane. The total number of TH-stained neurons in the SN pars compacta was calculated using the formula described. 48
Statistical analysis
The group means were compared using the Tukey’s post hoc test analysis for multiple groups. Differences were considered statistically significant at p < 0.05. All statistical analyses were performed using a commercially available software (SPSS Inc., version 12.0). All experiments were replicated independently 3–4 times.
Results
UA enhances glycolysis by upregulating PKM and LDH in MSCs
To demonstrate whether MSC priming with UA relies on glycolysis, we analyzed the expression levels of rate-limiting enzymes by quantitative RT-PCR. Glucose metabolism consists of interconnected pathways, with key junctions between glycolysis and the pentose phosphate pathway (PPP). Compared with the control MSCs, MSCs treated with either 200 μM or 400 μM UA led to increased mRNA levels of glycolysis enzymes, such as hexokinase 2 (HK2), 6-phosphofructo-2-kinase/fructose-2,6-biphosphatase 3 (PFKFB3), pyruvate kinase muscle isozyme (PKM), fructose-bisphosphate aldolase (ALDOA), and lactate dehydrogenase (LDH) (Figure 1(b)–(e)). mRNA levels of HK1 were comparable between UA-treated and control MSCs (Figure 1(a)). However, the mRNA levels of PPP enzymes, such as glucose 6-phosphate dehydrogenase (G6PD), 6-phosphogluconate dehydrogenase (PGD), and transketolase (TKT), showed no significant differences between UA-treated and control MSCs (Supplemental Figure 1). Additionally, we examined the concentration of L-lactate, the end product of glycolysis, initial step of which is the conversion of pyruvate to L-lactate by LDH. The L-lactate levels were increased in the UA-treated MSCs compared to those in control MSCs, indicating aerobic glycolysis triggered by UA (Figure 1(g)). These results showed that UA treatment could stimulate glycolysis by upregulating glycolysis enzymes in MSCs.

UA enhances glycolysis by upregulating PKM and LDH in MSCs: (a)–(f) Quantitative RT-PCR showed that UA-treated MSCs led to increased mRNA levels of glycolysis enzymes, such as HK2, PFKFB3, PKM, ALDOA, and LDH, compared to the control MSCs. (g) The L-lactate levels were increased in the UA-treated MSCs compared to those in control MSCs.
UA regulates self-renewal and differentiation potential of MSCs
Next, we examined whether upregulation of glycolysis promotes stemness in UA-treated MSCs. The stem cell properties of MSCs are characterized by their self-renewal ability, high proliferation rate, and multipotent differentiation into osteocytes, adipocytes, and chondrocytes. Both control MSCs and UA-treated MSCs expressed CD44 and CD105, positive MSC markers, but did not express CD34 and CD45, negative MSC markers (Figure 2(a)). We performed an MTS assay to examine whether UA treatment enhances the proliferation properties of the MSCs. UA-treated MSCs showed a significant increase in cellular density compared with control MSCs in a time-dependent manner, regardless of the UA concentration (Figure 2(b)). Thereafter, to evaluate UA impact on senescence, we analyzed the expression levels of p16 and p21. Expression levels of p16 increase after senescence, whereas p21 expressions rapidly increase in cells approaching replicative senescence. 49 The MSCs were treated with 200 μM or 400 μM UA, each for 24 h and 48 h. We found that the expression levels of p16 and p21 did not increase at an UA concentration of 200 μM after 24 h. However, the expression levels of p16 were increased with an UA concentration of 400 μM UA after 24 h or any UA concentration after 48 h. Expression levels of p21 also showed a similar tendency of increased expression, according to UA concentration and incubation time (Figure 2(c)). These results suggested that UA affects cellular senescence at high concentrations and after long incubation times. Next, we examined mRNA and protein levels of transcriptional factors responsible for stemness regulation, including OCT4, NANOG, and SOX2. Expression levels of OCT4 increased significantly after 24 h of both 200 and 400 μM UA treatment, whereas they decreased after 48 h, regardless of the UA concentration. Similarly, Expression levels of NANOG significantly increased after 24 h of 200 and 400 μM UA treatment and decreased after 48 h incubation, regardless of the UA concentration. Additionally, Expression levels of SOX2 increased after 24 h of incubation, whereas they markedly decreased after 48 h incubation, independent of UA concentrations (Figure 2(d)). This indicates that UA enhances stemness in conditions of short incubation and relatively low UA concentrations. Next, to test whether UA-induced stemness augmentation would result in increased differentiation potential of MSCs, we examined the mRNA levels of genes associated with differentiation, including osteogenesis (RUNX2, ALP), adipogenesis (PPARG, RHOA), and chondrogenesis (HAT1, BMP4). MSCs were treated with 200 μM or 400 μM UA, each for 24 h and 48 h and then, cultured without differentiation medium. UA treatment did not affect the expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes in MSCs (Supplemental Figure 2(a)). Additionally, we performed alizarin red S (ARS), oil red O (ORO), and alcian blue (AB) staining to identify osteogenic, adipogenic, and chondrogenic potential of MSCs, respectively. Immunostaining showed that UA treatment did not affect MSC differential potential toward any lineage (Supplemental Figure 2(b)). Moreover, MSCs were treated with 200 μM or 400 μM UA, each for 24 h and 48 h and then, induced with osteogenic or chondrogenic differentiation medium for 2 weeks and adipogenic differentiation medium for 3 weeks. They exhibited increased expression levels of adipogenesis-, osteogenesis- and chondrogenesis-related genes compared with naïve MSCs at an UA concentration of 200 and 24 h (Figure 2(e)). Additionally, immunostaining revealed that UA-treated MSCs after induction with a differentiation medium markedly increased their differentiation potential for osteogenesis, adipogenesis, and chondrogenesis (Figure 2(f)). Finally, we evaluated mRNA and protein levels whether MSC multipotency was sustained after removal of the UA-containing medium 24 h after UA initiation. Expression levels of OCT4, NANOG, and SOX2 that were significantly increased after 24 h of UA treatment were dramatically decreased after UA removal (Figure 2(g)). These data suggest that UA enhances stemness with a strict senescence regulation, depending on its concentration and incubation time.
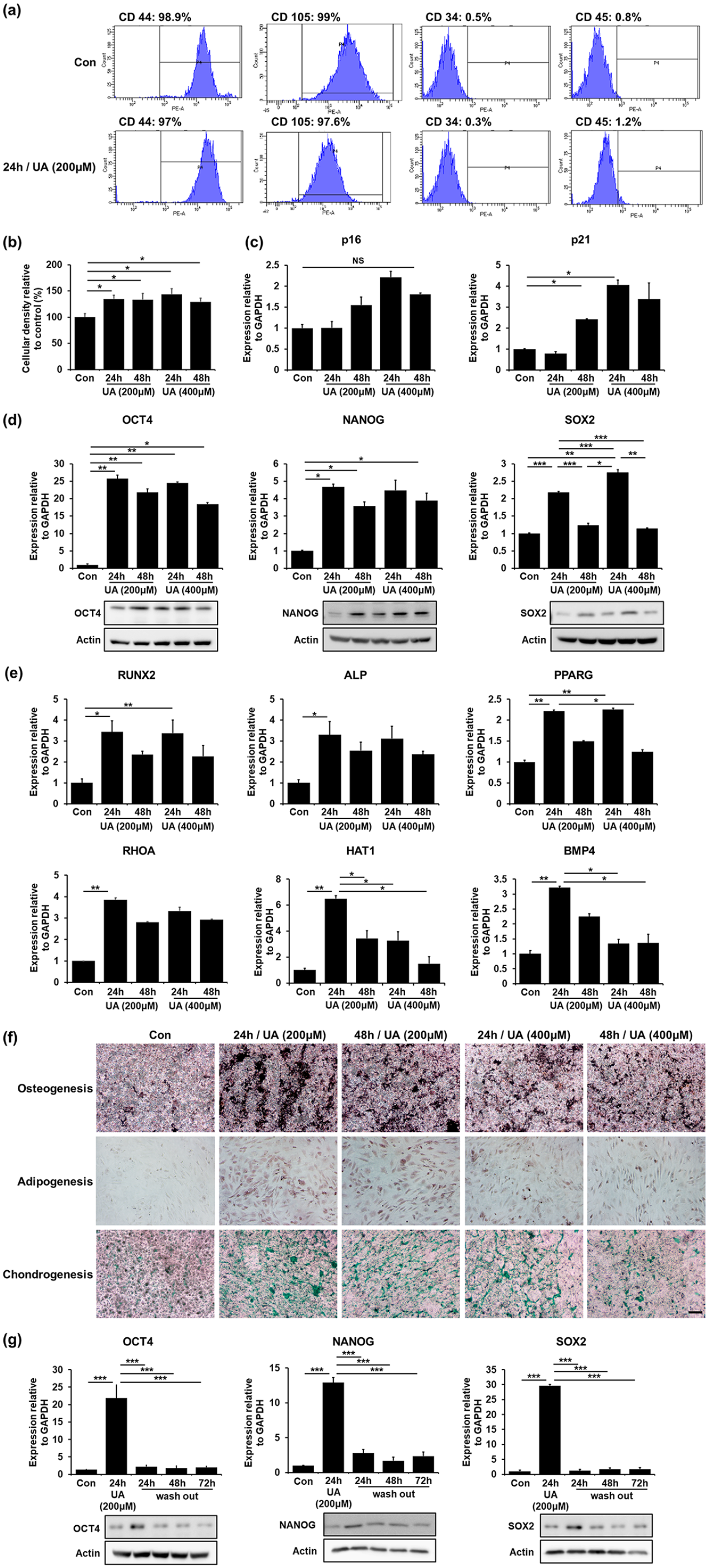
UA regulates self-renewal and differentiation potential of MSCs: (a) FACS analysis revealed that both control MSCs and 200 μM UA-treated MSCs expressed CD44 and CD105, but did not express CD34 and CD45, (b) MTS analysis showed that UA-treated MSCs significantly increased a cell density compared to control MSCs in a time-dependent manner, regardless of UA concentration, (c) Quantitative RT-PCR showed that the expression levels of the senescence markers p16 and p21 did not increase after 24 h incubation with 200 μM UA, (d) Quantitative RT-PCR and western blotting showed that the expression levels of the stemness markers OCT4, NANOG, and SOX2 increased after 24 h incubation with 200 μM UA, (e) Quantitative RT-PCR showed that UA-treated MSCs exhibited increased expression levels of osteogenesis (RUNX2, ALP), adipogenesis (PPARG, RHOA), and chondrogenesis (HAT1, BMP4)-related genes compared to naïve MSCs after induction with a differentiation medium, (f) Immunostaining showed that UA-treated MSCs markedly increased differentiation potential of MSCs toward osteogenesis, adipogenesis, and chondrogenesis after induction with a differentiation medium. Scale bar represents 200 μm, and (g) Quantitative RT-PCR and western blotting showed that expression levels of OCT4, NANOG, and SOX2 that were significantly increased after 24 h h of 200 μM UA treatment were dramatically decreased after UA removal.
MiR-137 regulates UA-associated MSC priming
Based on known miRNAs regulating self-renewal in stem cells, we screened 20 miRNAs to uncover possible UA-related stemness-controlling ones (Figure 3; Supple-mental Figure 3 and Table 1). We found that expression levels of miR-137 and miR-145 were tended to be decreased in UA-treated MSCs compared to those in control MSCs. Moreover, mRNA levels of miR-137 levels were markedly decreased in the UA-treated MSCs even compared to those of miR-145 (Figure 3(a) and (b)). The expression levels of the other miRNAs did not differ between UA-treated and control MSCs. These results suggest miR-137 as a candidate regulator of UA-associated MSC priming.

MiR-137 regulates UA-associated MSC priming. (a and b) Quantitative RT-PCR for self-renewal related miRNA showed that miR-137 and miR-145 expression levels were decreased in UA-treated MSCs compared to those in control MSCs.
Priming MSC with UA exerts neuroprotection in MPP+-treated SH-SY5Y cells
To examine the effects of UA-associated MSC priming on cell viability, SH-SY5Y cells were treated with MPP+ for 24 h. Then, the cells were co-cultured with control MSCs, primed MSCs, or primed MSCs with a 24 h -UA wash out. MPP+ treatment led to a significant decrease in cell survival compared to the control group. However, co-culture of MPP+-treated cells with control MSCs, primed MSCs, or primed MSCs with UA wash out in Transwell chambers led to increased cell viability compared to the MPP+-treated group. In addition, cell viability was significantly increased in the co-culture group with primed MSCs compared to that with control MSCs or primed MSCs with UA wash out (Figure 4(a)), indicating that UA-primed MSCs has a protective effect. MPP+ treatment led to a significant increase in ROS levels, compared to the control group (Figure 4(b)). However, co-culture of MPP+-treated cells with control MSCs, primed MSCs, or primed MSCs with UA wash out inhibited ROS generation compared to the MPP+-treated group (Figure 4(b)). In addition, MPP+ treatment significantly increased LDH release compared to the control group, whereas co-culture of MPP+-treated cells with control MSCs, primed MSCs, or primed MSCs with UA wash out significantly decreased the LDH release, compared to the MPP+-treated group (Figure 4(c)). The modulating effect on ROS and LDH production was more prominent with the primed MSCs than with the control or primed MSCs with UA wash out. Apoptosis analysis revealed that the co-culture with primed MSCs significantly attenuated the expression of cleaved caspase-3, cytochrome c, and Bax and increased Bcl-2 expression compared to the co-culture with control MSCs or primed MSCs with UA wash out (Figure 4(d)–(g)). These results demonstrate that priming MSCs inhibits apoptosis and increases neuronal survival in neurotoxin-treated cells.
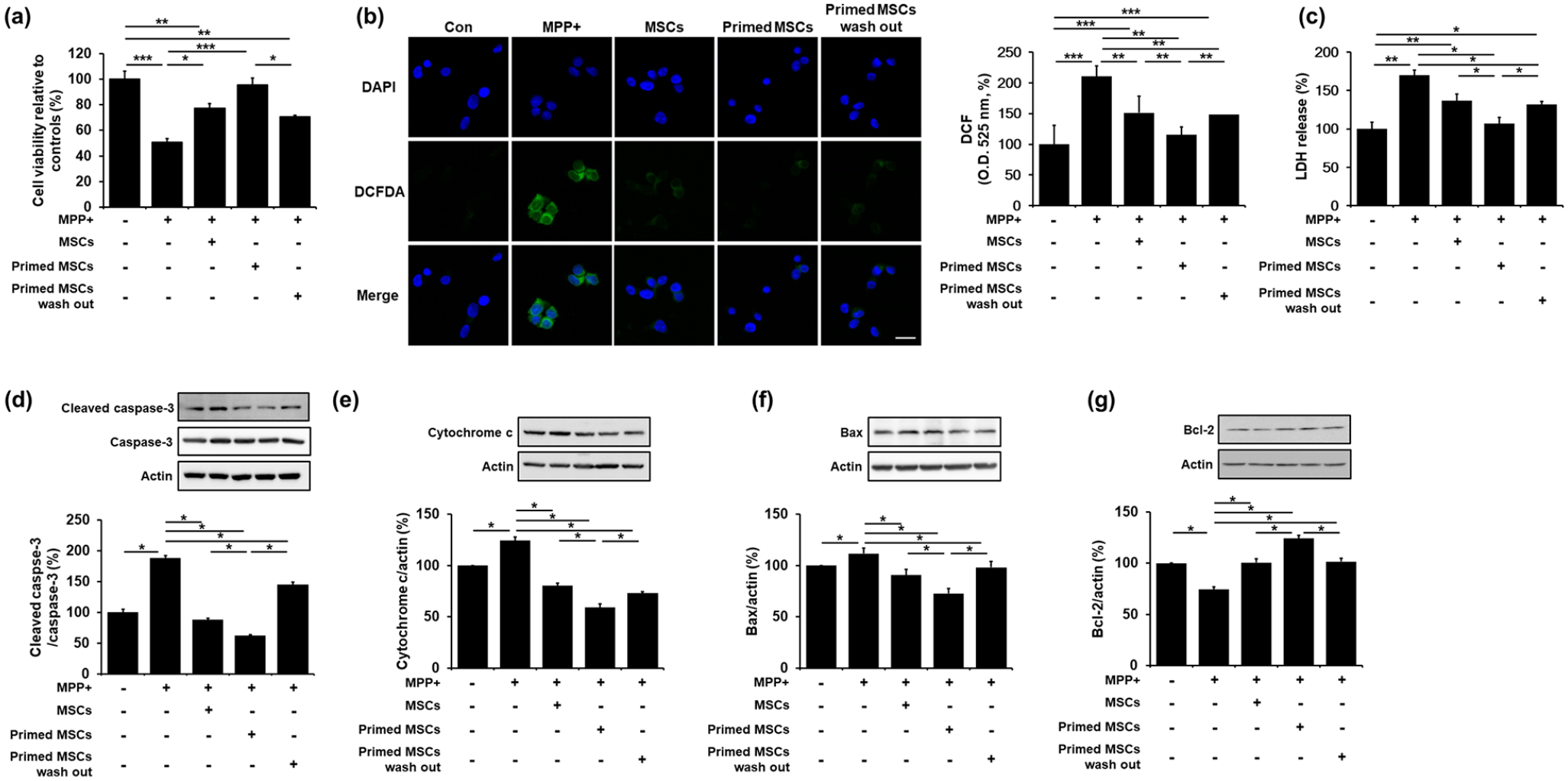
Priming MSC with UA exerts neuroprotection in MPP+-treated SH-SY5Y cells: (a) Cell viability showed that co-culture of MPP+-treated cells with primed MSCs resulted in significantly increased cell viability compared to the MPP+-treated, co-cultured with control MSCs, and co-cultured with primed MSCs with UA wash out groups. (b) ROS activity showed that co-culture of MPP+-treated cells with primed MSCs significantly inhibited ROS generation compared to the MPP+-treated, co-cultured with control MSCs, and co-cultured with primed MSCs with UA wash out groups. Scale bar represents 20 μm, (c) LDH cytotoxicity assay showed that co-culture of MPP+-treated cells with primed MSCs significantly inhibited ROS generation compared to the MPP+-treated, co-cultured with control MSCs, and co-cultured with primed MSCs with UA wash out groups. (d)–(g) Western blotting for apoptosis markers showed that co-culture with primed MSCs significantly attenuated cleaved caspase-3, cytochrome c, and Bax, and increased Bcl-2 expression compared to the co-culture with control or primed MSCs with UA wash out.
UA-enhancement therapy augments self-renewal and differentiation potential of mouse BM-MSCs
To assess whether UA-enhancement therapy increases self-renewal and differentiation potential of mouse BM-MSCs, we injected the mice with potassium oxonate (PO, an uricase inhibitor) and inosine 5′-monophosphate disodium salt hydrate (IMP, a precursor of UA) for 4 weeks. FACS analysis revealed that the MSCs isolated from control mice and PO- and IMP-treated mice expressed CD44 and CD105, positive MSC markers, but did not express CD34 and CD45, negative MSC markers (Figure 5(a)). Serum UA levels were significantly higher in PO- and IMP-treated mice and only PO-treated mice than control mice, but the levels were higher in PO- and IMP-treated mice than in only PO-treated mice (Figure 5(b)). The MTS assay showed that the MSC proliferation rate was significantly higher in PO- and IMP-treated mice than in the control or only PO-treated mice (Figure 5(c)). In addition, we examined the expression levels of p16, p21, OCT4, NANOG, and SOX2 to evaluate whether PO or IMP treatment regulated MSC senescence and stemness. The expression levels of p16 and p21 in PO- and IMP-treated mice did not differ from those in control or only PO-treated mice (Figure 5(d)). However, the expression levels of OCT4, NANOG, and SOX2 were significantly increased in PO- and IMP-treated mice compared to those in control or only PO-treated mice (Figure 5(e)). Furthermore, the expression levels of these stemness-associated markers were significantly increased in PO-treated mice compared to those in control mice (Figure 5(e)). Similarly, the mRNA levels of genes associated with osteogenesis (RUNX2, ALP), adipogenesis (PPARG, RHOA), and chondrogenesis (HAT1, BMP4) were increased in PO- and IMP-treated mice compared to those in control or only PO-treated mice (Figure 5(f)). Immu-nostaining showed that the differentiation potential toward osteogenesis, adipogenesis, and chondrogenesis was markedly increased in PO- and IMP-treated mice compared to that in control or only PO-treated mice (Figure 5(g)). Next, we examined expression levels of miR-137 and miR-145 to assess whether it affected by UA-enhancement therapy. The expression levels of miR-137 and miR-145 were relatively reduced in PO- and IMP-treated mice compared to those in control or only PO-treated mice (Figure 5(h)). These data suggest that UA-enhancement in mice induces MSC higher proliferation rate and stemness.

UA-enhancement therapy augments self-renewal and differentiation potential of mouse BM-MSCs. (a) FACS analysis showed that the control, and PO- and IMP-treated mice MSCs expressed CD44 and CD105, but did not express CD34 and CD45. (b) UA serum levels were significantly higher in PO- and IMP-treated mice than in control or only PO-treated mice. (c) MTS assay showed that MSC proliferation rate was significantly higher in PO- and IMP-treated mice than in control or only PO-treated mice. (d) Western blotting showed that p16 and p21 expression levels in PO- and IMP-treated mice did not differ from those in control or only PO-treated mice. (e) Western blotting showed that OCT4, NANOG, and SOX2 expression levels were significantly increased in the PO- and IMP-treated group compared to the control or only PO-treated mice. (f) Quantitative RT-PCR for differentiation markers showed that the expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes were increased in PO- and IMP-treated mice compared to those on control or only PO-treated mice. (g) Immunostaining showed that the differentiation potential of MSCs toward osteogenesis, adipogenesis, and chondrogenesis was markedly increased in the PO- and IMP-treated group compared to the control or only PO-treated group. Scale bar represents 200 μm, and (h) Quantitative RT-PCR showed that miR-137 and miR-145 expression levels were decreased in PO- and IMP-treated mice compared to those in control or only PO-treated mice.
MSC priming enhances neuroprotective properties in MPTP-treated mice
To investigate the potential neuroprotective effects of MSC priming, we performed a comparative analysis of the SN dopaminergic neurons of MPTP-treated parkinsonian mice with different MSC priming conditions. Immunohisto-chemical analysis revealed a significant decrease in the number of TH-positive neurons in MPTP-treated mice compared to control mice. Any type of MSC treatment increased the survival of tyrosine TH-positive neurons compared to the MPTP-treated group. Mouse or human MSC priming yielded much higher numbers of TH-positive neurons compared to either the MPTP-treated or the mouse control MSC group (Figure 6(a)). Consequently, mouse or human MSC priming led to significantly decreased expression levels of cleaved caspase-3 compared to either the MPTP-treated or the mouse control MSC group. Human MSC priming seemed to have a stronger neuroprotective capacity with respect to mouse MSC priming, with the human MSC priming treatment significantly decreasing the expression levels of cleaved caspase-3 (Figure 6(b)). In addition, we measured the levels of inflammatory cytokines to evaluate the immunomodulatory effects of MSC priming. The levels of pro-inflammatory cytokines, such as interleukin (IL)-1A, IL-17A, and interferon gamma (IFNγ) in MPTP-treated mice were increased compared to those in control mice, whereas any type of MSC treatment significantly decreased pro-inflammatory cytokine levels in MPTP-treated animals. Similarly, any type of MSC treatment significantly increased the levels of anti-inflammatory cytokines, such as IL-4, IL-10, and IL-11 in MPTP-treated mice (Figure 6(c)). The cytokine modulating effect was more prominent with mouse or human MSC priming than with mouse control MSC treatment. Consequently, both mouse and human MSC priming treatment led to the restoration of impaired motor coordination and balance on the Rotarod test compared to the control MSC group. Furthermore, human MSC priming showed a higher restorative capacity of impaired motor coordination compared with mouse MSC priming (Figure 6(d)).

MSC priming enhances neuroprotective properties in MPTP-treated mice. (a) TH-positive neuron counting in the SN at 4 weeks revealed that both mouse and human primed MSC treatment induced much higher numbers of dopaminergic neurons compared to both the MPTP-treated and the mouse control MSC group. Scale bar represents 200 μm. (b) Western blotting showed that both mouse and human primed MSC treatment significantly decreased cleaved caspase-3 expression levels compared to both the MPTP-treated and mouse control MSC group. (c) Quantitative RT-PCR showed that both mouse and human primed MSC treatment significantly decreased pro-inflammatory cytokine and increased anti-inflammatory cytokine expression levels compared to both the MPTP-treated and mouse control MSC group. (d) Both mouse and human primed MSC treatment led to the restoration of impaired motor coordination and balance in the Rotarod test compared to the control MSC group.
Discussion
In this study, we investigated the effects of UA on MSC stemness and its neuroprotective action in parkinsonian models. The major findings are as follows: (1) UA treatment in naïve MSCs stimulated glycolysis and upregulated transcriptional factors responsible for regulation of stemness, which increased the expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes. (2) Priming MSCs with UA increased cellular viability in neurotoxin-treated neuronal cells, relative to naïve MSCs, by inhibiting apoptotic signaling pathways. (3) MiR-137 markedly decreased in the UA-treated MSCs even compared to those of miR-145, suggesting that MiR-137 may regulate UA-associated MSC priming. (4) In an MPTP-treated animal model, the prosurvival effect of MSCs on dopaminergic neurons was prominent, with better behavioral recovery of motor deficits in primed MSC-treated mice compared to that in naïve MSC-treated mice. These data suggest that MSC priming with UA can provide a strategy to improve MSC application to the treatment of parkinsonian disorders.
UA is a final enzymatic product of purine nucleoside degradation in humans. High concentrations of circulating UA were proposed to be the major plasma antioxidant, protecting the cells from oxidative damage. Given that the balance between stem cell self-renewal and differentiation is critical for tissue homeostasis, oxidative stress resulting from excessive ROS production and impaired antioxidant systems can affect proliferation, differentiation, genomic mutations, aging, and death of stem cells.29,40,44,50 Additionally, evidence suggests that redox homeostasis plays a central role in maintaining stemness40,51 and reducing stem cell senescence. 43 Thus, disturbances of these mechanisms though excessive ROS production could lead to oxidative stress, resulting in stem cell dysfunction, such as senescence and loss of stemness during long-term expansion. In mammals, stem cells maintain low ROS levels to preserve their stemness and remain quiescent. However, research on the link between UA and stemness for clinical applications in neurodegenerative diseases is lacking.
During development, cell fate is defined by transcription factors that act as molecular switches to activate or repress specific gene expression programs. OCT4, NANOG, and SOX2 are considered to form a transcriptional regulatory circuitry for pluripotency and self-renewal of stem cells, including MSCs.36,52 OCT4 is the most upstream gene in the signaling pathway that regulates MSC self-renewal.52–54 Thus, the metabolism of undifferentiated MSCs during proliferation is primarily associated with glycolysis under both anaerobic and aerobic conditions, whereas mitochondrial OXPHOS is associated with differentiated MSCs and loss of multipotency.30,44 In the present study, we found that UA treatment stimulated glycolysis by upregulating PKM and LDH, with no changes in PPP enzymes. Specifically, glycolysis-related enzymes, such as PFKFB3, PKM, ALDOA, and LDH were significantly upregulated in UA-treated MSCs. In addition, low-concentration and short-incubation UA treatment did not affect MSC senescence, which was accompanied by a significant increase in MSC proliferation properties. Expression levels in OCT4, NANOG, and SOX2, stemness-related markers, in MSCs were increased by low-concentration and short-incubation UA treatment. However, these conditions did not affect the mRNA levels of genes associated with differentiation, including osteogenesis, adipogenesis, and chondrogenesis, suggesting no impact on the differentiation potential of MSC. UA treatment in lineage-specific differentiated MSCs, nevertheless, exhibited increased expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes, indicating an effect on the differentiation potential of MSC. Moreover, our in vivo data further confirmed the impact of UA on MSC stemness modulation. Mice undergoing UA-enhancement therapy showed increased MSC proliferation, and MSCs in IMP- and PO-treated mice exhibited increased expression of OCT4, NANOG, and SOX2, with no changes related to cellular senescence. Accordingly, our results suggest that UA enhances MSC stemness, with a strict regulation of senescence, possibly depending on UA concentration and incubation time.
MSC priming with UA exerted neuroprotection in neurotoxin-induced parkinsonian cellular and animal models. UA-primed MSCs significantly increased cell viability in neurotoxin-treated neuronal cells compared to naïve MSCs by inhibiting apoptotic signaling pathways, with concomitant restoration of neurotoxin-induced distorted oxidative stress. In the animal study, the protective effect of MSCs on dopaminergic neurons in MPTP-treated mice was prominent, regardless of the MSC primed or naïve state. However, the primed MSCs led to the rescue of more dopaminergic neurons in the SN compared with naïve MSCs in the MPTP-treated mice. In addition, mice treated with primed MSCs had decreased expression levels of cleaved caspase-3 in the midbrain compared to the mice receiving naïve MSCs. Consequently, motor recovery was prominent in MPTP-treated mice receiving both primed and naïve MSCs, but with a tendency of a better behavioral outcome with the primed MSCs. In terms of species-specific differences in the primed MSCs, human MSCs appeared to have more neuroprotective effects than mouse MSCs. Despite many similarities, MSC species variability affects not only the functional expression of various chemokine receptors but also other MSC behaviors. 55 It is possible that species-specific differences may influence MSC priming; however, further studies are needed to better inform the evidence for this issue.
Evidence has demonstrated that miRNAs are involved in apoptosis, 56 self-renewal, 57 and differentiation 58 of MSCs. In this study, we found decreased expression levels of miR-137 and miR-145 in UA-treated MSCs, suggesting that they could promote MSC stemness. Especially, mRNA levels of miR-137 were more profoundly decreased in the UA-treated MSCs even compared to those of miR-145 levels. Several studies reported decreased expression of miR-137 can lead to increased levels of stemness-related transcriptional factors, including OCT4, NANOG, SOX2, and KLF4,37,59 whereas its overexpression induces reduced neural stem cell proliferation, with enhanced neuronal and glial differentiation. 60 miR-137 also affects cancer stem cell self-renewal, and its downregulation significantly promoted stemness by targeting KLF12. 61 Furthermore, miR-137 seems to have a critical role in MSC proliferation and differentiation. 61 In addition, miR-145 expression was known to be low in self-renewing human embryonic stem cells, but it was highly upregulated during differentiation by regulating OCT4, SOX2, and KLF4. 62 Taken together, these results suggest that miR-137 and miR-145 may be closely coupled with MSC stemness regulation by UA.
Conclusions
The present data suggest that MSC priming with UA exerts neuroprotective effects through enhanced stemness and differentiation potential in parkinsonian models (Figure 7). Therefore, this could provide a novel strategy to improve MSC use for the treatment of parkinsonian disorders.
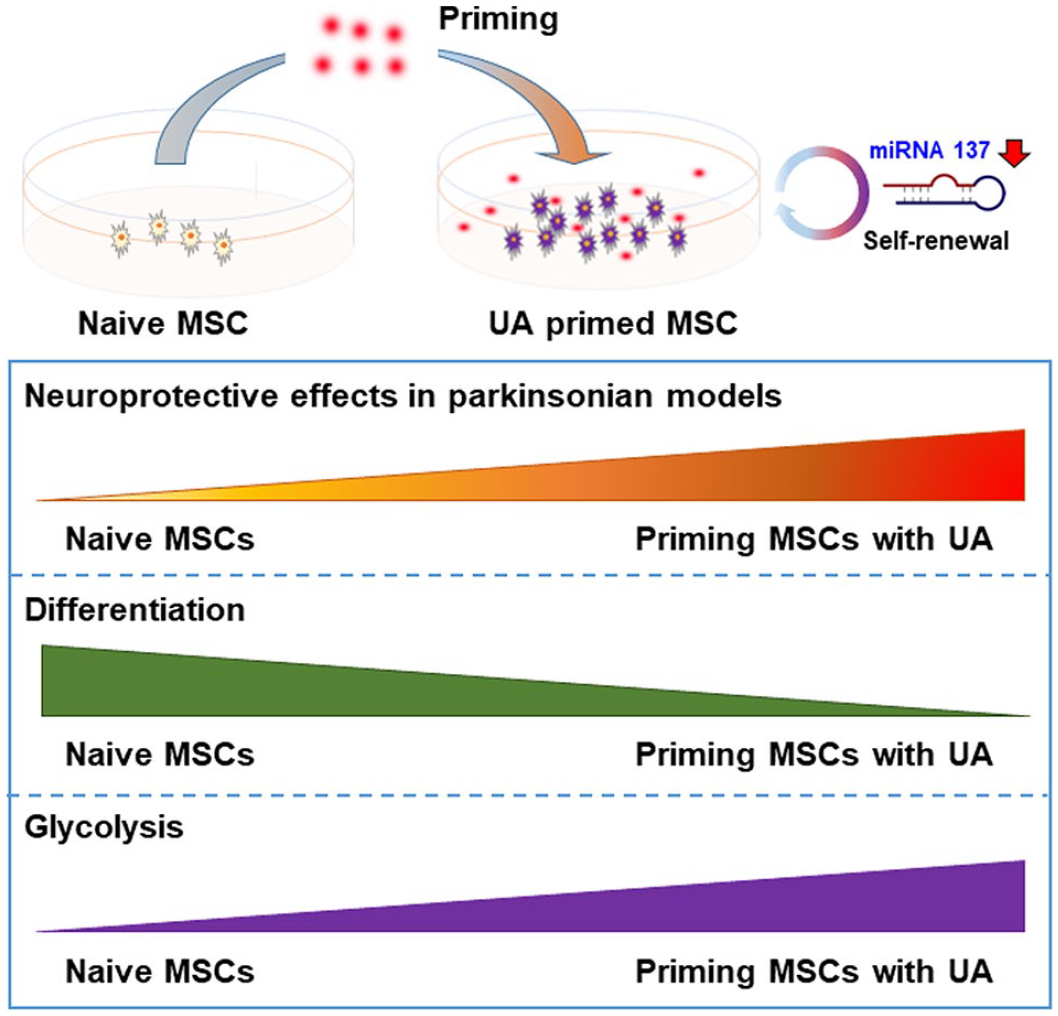
A schematic illustration of the biological action and mechanistic role of UA-priming of MSCs in parkinsonian models. UA treatment in naïve MSCs stimulated glycolysis and upregulated transcriptional factors responsible for regulation of stemness, which increased the expression levels of osteogenesis-, adipogenesis-, and chondrogenesis-related genes via modulation of MiR-137. Priming MSCs with UA increased neuroprotective properties relative to naïve MSCs. These data suggest that MSC priming with UA can provide a strategy to improve MSC application to the treatment of parkinsonian disorders.
Supplemental Material
sj-docx-1-tej-10.1177_20417314211004816 – Supplemental material for Priming mesenchymal stem cells with uric acid enhances neuroprotective properties in parkinsonian models
Supplemental material, sj-docx-1-tej-10.1177_20417314211004816 for Priming mesenchymal stem cells with uric acid enhances neuroprotective properties in parkinsonian models by Ha Na Kim, Jin Young Shin, Dong Yeol Kim, Ji Eun Lee and Phil Hyu Lee in Journal of Tissue Engineering
Footnotes
Authors’ contributions
H.N.K.: conception and design, collection and/or assembly of data, manuscript writing, final approval of manuscript; J.Y.S., D.Y.K., and J.E.L.: technical assistance, final approval of manuscript; P.H.L.: supervision of study, data analysis and interpretation, financial support, final approval of manuscript.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics approval
All experimental procedures were conducted in accordance with the relevant ethical guidelines and regulations approved by the Institutional Animal Care and Use Committee (IACUC) at the Yonsei University Health System.
Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This research was supported by the Basic Science Research Program through the National Research Foundation of Korea (NRF), funded by the Ministry of Science, ICT and Future Planning (NRF2019R1A2C208546212), and the Brain Korea 21 PLUS Project for Medical Science, Yonsei University.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.