
Abstract
Silk fibroin scaffolds were investigated for their ability to support attachment, proliferation, and differentiation of human gastrointestinal epithelial and smooth muscle cell lines in order to ascertain their potential for tissue engineering. A bi-layer silk fibroin matrix composed of a porous silk fibroin foam annealed to a homogeneous silk fibroin film was evaluated in parallel with small intestinal submucosa scaffolds. AlamarBlue analysis revealed that silk fibroin scaffolds supported significantly higher levels of small intestinal smooth muscle cell, colon smooth muscle cell, and esophageal smooth muscle cell attachment in comparison to small intestinal submucosa. Following 7 days of culture, relative numbers of each smooth muscle cell population maintained on both scaffold groups were significantly elevated over respective 1-day levels—indicative of cell proliferation. Real-time reverse transcription polymerase chain reaction and immunohistochemical analyses demonstrated that both silk fibroin and small intestinal submucosa scaffolds were permissive for contractile differentiation of small intestinal smooth muscle cell, colon smooth muscle cell, esophageal smooth muscle cell as determined by significant upregulation of α-smooth muscle actin and SM22α messenger RNA and protein expression levels following transforming growth factor-β1 stimulation. AlamarBlue analysis demonstrated that both matrix groups supported similar degrees of attachment and proliferation of gastrointestinal epithelial cell lines including colonic T84 cells and esophageal epithelial cells. Following 14 days of culture on both matrices, spontaneous differentiation of T84 cells toward an enterocyte lineage was confirmed by expression of brush border enzymes, lactase, and maltase, as determined by real-time reverse transcription polymerase chain reaction and immunohistochemical analyses. In contrast to small intestinal submucosa scaffolds, silk fibroin scaffolds supported spontaneous differentiation of esophageal epithelial cells toward a suprabasal cell lineage as indicated by significant upregulation of cytokeratin 4 and cytokeratin 13 messenger RNA transcript levels. In addition, esophageal epithelial cells maintained on silk fibroin scaffolds also produced significantly higher involucrin messenger RNA transcript levels in comparison to small intestinal submucosa counterparts, indicating an increased propensity for superficial, squamous cell specification. Collectively, these data provide evidence for the potential of silk fibroin scaffolds for gastrointestinal tissue engineering applications.
Introduction
The gastrointestinal (GI) tract is a continuous system of tubular organs, including the esophagus, stomach, small intestine, and colon, which is responsible for the transport and digestion of food, absorption of nutrients, and excretion of waste. A wide spectrum of benign and malignant GI pathologies frequently requires surgical intervention to repair or replace damaged tissues in order to preserve alimentary function. In patients afflicted with short bowel syndrome (SBS) wherein a reduction of more than 70% of normal jejunal–ileal length has occurred, 1 intestinal lengthening procedures 2 and allogeneic intestinal transplantation 3 have been deployed to increase functional absorptive capacity. However, major drawbacks with these approaches exist, including the continued reliance on total parenteral nutrition following intestinal elongation4,5 as well as immunojection risks in individuals receiving allografts.6,7 Long-gap esophageal defects resulting from esophageal atresia, strictures, and squamous cell carcinoma are often repaired with gastric pull-up or interposition grafts using either jejunum or colon.8,9 Unfortunately, these methods are associated with substantial reductions in esophageal motility as well as donor site morbidity10,11 which can severely impair patient quality of life.12,13 Surgical colon resection is frequently utilized for management of disorders of the large intestine such as Crohn’s and Hirschsprung’s diseases. 14 However, increased stool frequency and fecal incontinence represent undesirable consequences of this mode of therapy.15,16 Given the limitations associated with conventional surgical approaches, there exists a substantial need for the development of alternative strategies for GI reconstruction.
Tissue engineering strategies deploying three-dimensional (3D) biodegradable scaffolds either alone or seeded with primary or multi-potent cell sources have been investigated as implants for GI defect repair.17–19 Cell-free grafts composed of decellularized tissues such as small intestinal submucosa (SIS), acellular dermis, collagen-based sponges, and gastric acellular matrices have been shown to promote de novo epithelial and smooth muscle tissue formation in animal models of esophageal and small intestinal injury by supporting host tissue integration.18,20–22 However, deleterious side effects including graft contracture, implant perforation, obstruction, and stenosis are frequently observed,23–26 thus raising concerns over the translational potential of these biomaterial configurations. Synthetic polyester-based matrices seeded with organoid units, multi-cellular clusters of epithelium, and mesenchyme harvested from neonatal or postnatal GI tissue have been reported to encourage tissue regeneration in defects of the esophagus, 27 small intestine, 28 and colon. 29 Unfortunately, degradation metabolites of polyesters are known to elicit chronic inflammatory responses in vivo 30 and therefore have the potential to negatively impact long-term organ function due to adverse foreign body reactions. 31 In addition, the limited availability of human neonatal or postnatal donor tissue for organoid procurement represents a practical barrier for widespread clinical utilization of this technology. 32
We hypothesize that the ideal strategy for GI tissue reconstruction would consist of an “off-the-shelf” acellular implant with the structural, mechanical, and degradative properties necessary to provide initial reinforcement to defect sites while allowing for gradual remodeling, host tissue ingrowth, and subsequent maturation of site-appropriate, functional tissue in the absence of adverse inflammatory reactions. Biomaterials derived from Bombyx mori silk fibroin (SF) represent attractive candidates for GI tissue engineering due to their high structural strength and elasticity, 33 diverse processing plasticity, 34 tunable biodegradability,35,36 and low immunogenicity.31,33 Previous studies from our laboratory have demonstrated the feasibility of bi-layer SF scaffolds to serve as acellular, biodegradable matrices for functional tissue regeneration of bladder37–39 and urethral 40 defects. The unique architecture of the bi-layer SF scaffold configuration is also particularly suited for repair of GI perforations or replacement of diseased tissue sites since its porous compartment has the potential to promote ingrowth of surrounding host tissue while an SF film annealed to the porous layer is designed to provide a fluid-tight seal for retention of GI contents during defect consolidation.37,38 In the present study, we investigated the biocompatibility of this scaffold design for GI tissue engineering by evaluating its ability to support attachment, proliferation, and differentiation of human GI epithelial and smooth muscle cell (SMC) lines in vitro. The ability of biomaterials to support these cellular processes is crucial for promoting host tissue integration and functional maturation of regenerating tissue.
Materials and methods
Biomaterials
Bi-layer SF scaffolds were prepared using previously described procedures.37,40, 41 Briefly, cocoons from Bombyx mori were boiled for 20 min in an aqueous solution of 0.02 M Na2CO3, and rinsed with distilled water to eliminate sericin and other contaminating proteins. Purified SF was solubilized in a 9 M LiBr solution and dialyzed (Pierce, Woburn, MA) against distilled water for 4 days with volume changes every 8 h. The resultant aqueous SF solution was diluted with distilled water to 6%−8% wt/vol and utilized for scaffold fabrication. The SF solution (8% wt/vol) was poured into a rectangular casting vessel and dried in a laminar flow hood at room temperature for 48 h to achieve formation of a SF film. A 6% wt/vol SF solution was then mixed with sieved granular NaCl (500–600 µM, average crystal size) in a ratio of 2 g NaCl per mL of SF solution and layered on to the surface of the SF film. The resultant solution was allowed to cast and fuse to the SF film for 48 h at 37°C, and NaCl was subsequently removed by washing the scaffold for 72 h in distilled water with regular volume changes. The morphology of the bi-layer SF scaffold has been previously reported. 37 Briefly, the solvent-cast/NaCl-leached layer comprised the bulk of the total matrix thickness (2 mm) and resembled a foam configuration with large pores (pore size, ~400 µm) interconnected by a network of smaller pores dispersed along their periphery. This compartment was buttressed on the external face with a homogeneous, non-porous SF layer (200 µm thick) generated by film annealment during casting. Prior to in vitro experiments, bi-layer SF scaffolds were sterilized in 70% ethanol and rinsed in phosphate buffered saline (PBS) overnight. SIS matrices (Cook, Bloomington, IN) were evaluated in parallel as a standard point of comparison. Tensile properties of both scaffold configurations have been previously reported. 37
GI SMCs
Primary SMC (passage 1) derived from human small intestine SMC (siSMC), colon SMC (cSMC), and esophageal SMC (eSMC) (ScienCell Research Laboratories, Carlsbad, CA) were expanded for two additional passages on tissue culture plastic in SMC medium (SMCM, ScienCell) according to the manufacturer’s instructions. SMCM consists of a bicarbonate buffered basal medium containing 2% fetal bovine serum (FBS), 1% of SMC growth supplement (SMCGS), and 1% of penicillin/streptomycin solution (P/S). For cell attachment and proliferation analyses, SMC lines (passage 4) were independently seeded on bi-layer SF and SIS matrices (106 cells/scaffold) in SMCM and cultured statically in conical tubes for up to 7 days. Medium exchange was carried out every 3 days. To induce contractile differentiation, SMC lines were seeded on matrices as described above, serum depleted for 24 h in SMCM containing 0.5% fetal calf serum (FCS), and subsequently exposed to 2.5 ng/mL transforming growth factor-β1 (TGF-β1) (R&D Systems, Minneapolis, MN) for 24 h. Control cultures were maintained in parallel in SMCM for 3 days.
GI epithelial cells
The human T84 epithelial cell line derived from colonic adenocarcinoma (American Type Culture Collection (ATCC), Manassas, VA) was expanded to passage 56 on tissue culture plastic in complete growth medium consisting of a 1:1 mixture of Ham’s F12 medium and Dulbecco’s modified Eagle’s medium (DMEM), 2.5 mM L-glutamine, 15 mM hydroxyethyl piperazine ethanesulfonic acid (HEPES), 0.5 mM sodium pyruvate, and 1200 mg/L sodium bicarbonate (DMEM:F-12, ATCC), supplemented with 5% FBS (Life Technologies, Grand Island, NY). Human primary esophageal epithelial (eEP) cells (passage 1) (ScienCell) were expanded for three additional passages on tissue culture plastic in Epithelial Cell Medium-2 (EpiCM-2, ScienCell) according to the manufacturer’s instructions. EpiCM-2 consists of 1% of epithelial cell growth supplement-2 (EpiCGS-2) and 1% of P/S. eEP and T84 cells were independently seeded on bi-layer SF and SIS matrices (106 cells/scaffold) and cultured statically in conical tubes in respective growth media for up to 7 or 14 days to assess cell attachment, extent of proliferation, and differentiation status. Medium exchange was performed every 3 days.
Cell attachment and proliferation analyses
The relative number of metabolically active cells following 24 h of cell seeding and over the course of cultivation on each matrix group was determined by the alamarBlue assay (Life Technologies) according to the manufacturer’s instructions. Briefly, scaffolds seeded with cell lines were incubated in their respective culture medium supplemented with 10% alamarBlue reagent for 2 h at 37°C with 5% CO2. Post reaction medium aliquots (100 µL) were transferred to 96-well plates and quantified for fluorescence intensity within a FLUOstar Omega plate reader (BMG Labtech Inc., Durham, NC) using an excitation wavelength of 560 nm and an emission wavelength of 590 nm. Non-seeded matrices were screened in parallel as background controls. Relative cell numbers were calculated as the degree of relative fluorescence units (FU) per construct as previously described. 42
Messenger RNA analysis
Total RNA was extracted from cell seeded scaffolds according to the single step acid-phenol guanidinium method 43 using Trizol reagent (Life Technologies). Messenger RNA (mRNA) was enriched from total RNA using the RNeasy kit (Qiagen Inc., Valencia, CA), and complementary DNAs (cDNAs) were synthesized using the High-Capacity cDNA Transcription kit (Applied Biosystems, Foster City, CA) following the manufacturer’s instructions. Real-time reverse transcription polymerase chain reaction (RT-PCR) reactions were performed and analyzed using the Applied Biosystems StepOnePlus™ Real-time PCR Detection System and StepOne Software (version 2.1). cDNA samples were assessed for genes of interest and the housekeeping gene, GAPDH, in independent reactions using the Taqman Universal PCR master mix in combination with commercially available primers and probes consisting of Assays-on-Demand™ Gene Expression kits (Applied Biosystems) following the manufacturer’s instructions. Expression kits included α-smooth muscle actin (SMA), Hs00426835_g1; SM22α, Hs00162558_m1; lactase, Hs00158722_m1; maltase, Hs01090216_m1; cytokeratin (CK) 4, Hs00361611_m1; CK13, Hs00357961_g1; involucrin, Hs00846307_s1; GAPDH, Hs02758991_g1. For each cDNA sample, the threshold cycle (Ct) was defined as the cycle number at which amplification of the target gene was within the linear range of the reaction. Relative expression levels for each gene of interest were calculated by normalizing the target gene transcript level (Ct) to the respective GAPDH level with the maximum expression values per gene displayed at a 100 per condition as described previously. 42
Histological, immunohistochemical, and scanning electron microscopy analyses
Cell-seeded constructs were fixed in 10% neutral-buffered formalin, dehydrated in graded alcohols, and then embedded in paraffin. Sections (5 µm) were cut and then stained with hematoxylin and eosin (H&E) using routine histological protocols. For immunohistochemical (IHC) analyses, contractile smooth muscle markers such as α-SMA and SM22α and intestinal epithelial markers including brush border enzymes, lactase, and maltase were detected using the following primary antibodies: anti-α-SMA (Sigma-Aldrich, St. Louis, MO; 1:200 dilution), anti-SM22α (Abcam, Cambridge, MA; 1:200 dilution), anti-lactase (Abcam; 1:200 dilution), and anti-maltase (Santa Cruz Biotechnology, Dallas, TX; 1:200 dilution). Sections were then incubated with species-matched Cy3-conjugated secondary antibodies (EMD Millipore, Billerica, MA) and nuclei were counterstained with 4′,6-diamidino-2-phenyllindole (DAPI). Specimens were visualized using an Axioplan-2 microscope (Carl Zeiss MicroImaging, Thornwood, NY), and representative images were acquired using Axiovision software (version 4.8). In some cases, cell distribution in seeded constructs was determined by scanning electron microscopy (SEM) using previously published procedures. 44
Statistical analyses
All quantitative measurements were collected with N = 3–4 independent replicates per data point from one representative experiment and expressed as mean ± standard deviation. Data for these measurements were analyzed with the Mann–Whitney U test for independent samples and the Wilcoxon signed-rank test for paired samples using SPSS Statistics software v19.0 (http://www.spss.com). Statistically significant values were defined as p < 0.05.
Results and discussion
Bi-layer SF and SIS matrices were assessed for their ability to mediate attachment and proliferation of GI SMC lines (Figure 1). Following 24 h of cell seeding, alamarBlue analysis demonstrated that bi-layer SF scaffolds supported significantly higher levels of attachment for all SMC lines examined in comparison to SIS matrices. At the 7-day timepoint, relative numbers of each SMC population maintained on both bi-layer SF and SIS scaffolds were significantly elevated over their respective 1-day levels—indicative of cell proliferation. Parallel histological evaluations (hematoxylin and eosin (H&E) analysis) at 7 days revealed that each SMC line displayed a spindle-shaped morphology and was organized into multi-cellular layers primarily dispersed along the periphery of both biomaterial surfaces. Overall, these data show that bi-layer SF matrices are capable of promoting GI SMC attachment and proliferation.

Attachment and proliferation of GI SMC lines on scaffold groups. (a–c) AlamarBlue analysis of the extent of relative cell attachment and proliferation for each SMC line cultured independently on bi-layer SF and SIS matrices over the course of 7 days. FU = Fluorescence units, mean ± SD per data point. #p < 0.05, in comparison to levels observed on SIS constructs at 1 day of culture. *p < 0.05, in comparison to respective levels observed on SIS and bi-layer SF constructs at 1 day of culture. (a′–c′) Photomicrographs of SMC-seeded constructs (H&E-stained sections) following 7 days of culture as described in respective (a–c) counterparts. Scale bars = 200 µm. SC denotes scaffolds.
Phenotypic modulation of SMC from a proliferative, synthetic state into a quiescent, contractile phenotype is a dynamic, reversible process which plays key roles in tissue homeostasis and response to injury throughout hollow organs. 45 In order for biomaterial grafts to reconstitute the contractile properties of tissue defects and support peristalsis of the digestive tract, they must serve as permissive substrates for contractile differentiation of synthetic SMC. Following tissue isolation and ex vivo expansion, primary SMC populations are known to dedifferentiate from a contractile to a synthetic phenotype.46–47 Serum deprivation in combination with TGF-β1 treatment has been reported to induce re-acquisition of contractile markers in ex vivo expanded, synthetic SMC derived from vascular 48 and urogenital tissues.42,46 Therefore, we investigated the potential of these differentiation stimuli to elicit similar responses in GI SMC lines cultured on bi-layer SF and SIS matrices (Figure 2). Real-time RT-PCR analysis demonstrated that following TGF-β1 treatment, each SMC type cultured on both scaffold groups significantly upregulated α-SMA and SM22α mRNA transcript levels over respective control values. In addition, IHC evaluations revealed that all TGF-β1-treated constructs displayed prominent degrees of α-SMA and SM22α protein expression with qualitatively similar levels of positive staining observed across all cell types and matrix configurations examined. In contrast, non-stimulated control groups displayed qualitatively weak and sparse expression of both markers by comparison (data not shown). These results demonstrate that bi-layer SF matrices are efficacious in supporting contractile differentiation of GI SMC lines.

Contractile differentiation of GI SMC lines on matrix groups. (a–c) Real-time RT-PCR analyses of mRNA transcript levels of contractile markers (α-SMA and SM22α) in synthetic (SYN) and TGF-β1-treated constructs seeded with SMC lines. Levels normalized to GAPDH expression. Mean ± SD per data point. For each marker, *p < 0.05, in comparison to levels in respective SYN controls. (a′–c′) Photomicrographs of contractile protein expression in TGF-β1-treated constructs as described in respective (a–c) counterparts. Immunofluorescence of contractile proteins (Cy3 fluorophore, red). DAPI nuclear counterstain (blue). Scale bars = 100 µm.
The diverse epithelia of the GI tract serve a number of critical functions such as enzyme secretion, nutrient absorption, as well as participation in innate and adaptive immune responses. 49 The ability of tissue-engineered constructs to reconstitute the epithelium of GI tissue defects depends on the potential of scaffold configurations to promote host epithelial ingrowth and differentiation. Bi-layer SF and SIS matrices were first evaluated for their capacity to support attachment and proliferation of two GI epithelial cell lines: T84 and eEP (Figure 3). AlamarBlue analysis revealed that following initial cell seeding, both scaffold groups displayed similar levels of relative cell attachment for each cell line studied. Following 14 days of culture, significant increases in relative cell numbers were observed in both T84-seeded biomaterials over respective 1-day levels, and histological evaluations (H&E analysis) demonstrated the formation of polarized cell layers lining the perimeter of each scaffold configuration. In addition, relative cell numbers of eEP cells on both bi-layer SF and SIS matrices at 7 days of culture were also found to significantly increase with respect to 1-day values. However, in contrast to T84 cells, SEM analysis demonstrated that eEP cells were localized in disperse patches along each scaffold surface and did not form detectable cohesive cell layers as observed on other biomaterial substrates. 50 Our previous results have shown the ability of extracellular matrix coatings such as fibronectin to enhance urologic epithelial interactions to SF biomaterials. 42 This strategy may represent a useful approach in order to improve construct cellularity in the current system.

Attachment and proliferation of GI epithelial cell lines on scaffold groups. (a and b) AlamarBlue analysis of the extent of relative cell attachment and proliferation for each epithelial cell line cultured independently on bi-layer SF and SIS matrices over the course of 7 or 14 days. FU = Fluorescence units, mean ± SD per data point. *p < 0.05, in comparison to respective levels observed on SIS and bi-layer SF constructs at 1 day of culture. (a′ and b′) Photomicrographs of epithelial cell lines cultured on each scaffold group for 7 or 14 days (H&E-stained sections, T84 and SEM, eEP). For a′, scale bar = 200 µm. SC denotes scaffolds. For b′, scale bar = 30 µm.
T84 and eEP cell lines have been demonstrated to undergo spontaneous differentiation upon confluency in various two-dimensional (2D) and 3D cell culture models.50–53 T84 cells acquire features of small intestinal enterocytes including expression of the brush border enzymes such as lactase and maltase;51,52 while proliferating, CK5 + CK14 + basal eEP cells mature into a suprabasal cell lineage expressing CK4 + CK13 + a superficial, squamous cell phenotype which produces involucrin.50,53 The bi-layer SF and SIS scaffolds were assessed for their ability to support spontaneous differentiation of T84 and eEP cell lines (Figure 4). Real-time RT-PCR analysis demonstrated that T84 cells cultured on both scaffold groups significantly increased mRNA transcript levels of lactase and maltase at 14 days with respect to 1-day levels. In addition, IHC assessments revealed that T84 cells cultured on each matrix configuration for 14 days displayed qualitatively similar levels of lactase and maltase protein expression. These data show the ability of bi-layer SF scaffolds to support enterocytic differentiation of T84 cells to a comparable degree as SIS.
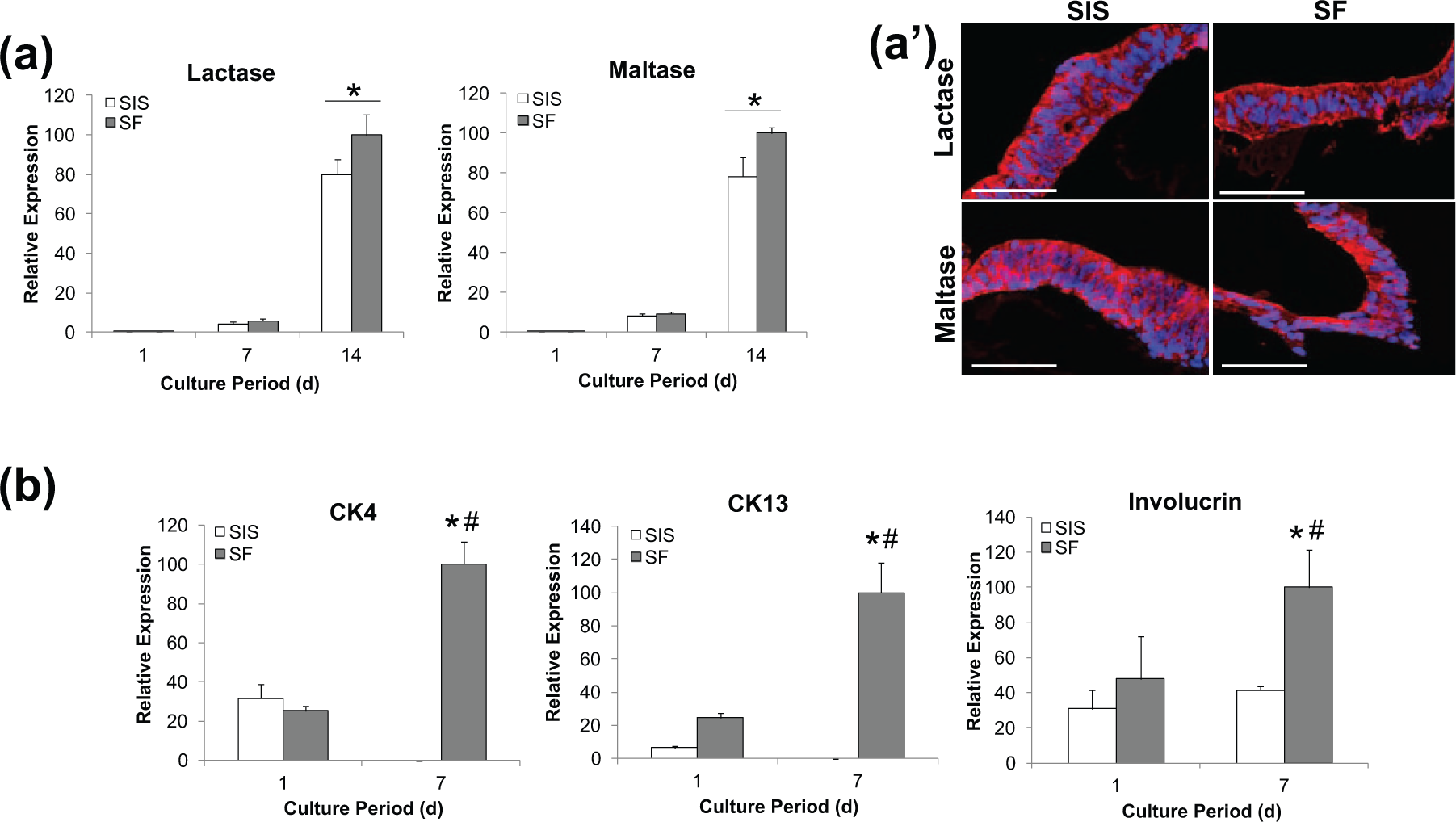
Differentiation of GI epithelial cell lines on matrix groups. Real-time RT-PCR analyses of mRNA transcript levels of (a) enterocyte markers (lactase and maltase) in T84 cells cultured on matrix groups over the course of 14 days and (b) markers of suprabasal (CK4, CK13) and superficial (involucrin) cell phenotypes in eEP cells cultured on scaffold groups over the course of 7 days. *p < 0.05, in comparison to respective levels observed on SIS and bi-layer SF constructs at 1 day of culture. #p < 0.05, in comparison to respective levels observed in SIS group at 7 days of culture. (a′) Photomicrographs of enterocyte protein T84-seeded constructs at 14 days of culture as described in respective (a) counterpart. Immunofluorescence of enterocyte proteins (Cy3 fluorophore, red). DAPI nuclear counterstain (blue). Scale bars = 100 µm.
In contrast to the results obtained with T84 cells, distinct differences were observed in the capacity of the biomaterial groups to promote eEP differentiation. Following 7 days of culture on bi-layer SF scaffolds, real-time RT-PCR analysis demonstrated that eEP cells significantly upregulated CK4 and CK13 mRNA transcript levels over 1-day values, indicative of suprabasal cell specification. However, these markers declined from 1-day baseline levels when eEP cells were cultured on SIS matrices. Evidence for superficial, squamous eEP differentiation was detected in both scaffold groups by the presence of involucrin expression; however, bi-layer SF constructs supported significantly higher mRNA transcript levels (2.4-fold) at 7 days of culture in comparison to SIS matrices. Bioactive growth factors in SIS scaffolds such as TGF-β1 54 have the potential to exert inhibitory roles in eEP cell differentiation processes, 55 and therefore, their presence may account for the observed differences between the two matrix configurations. Indeed, previous studies have reported incomplete epithelialization of SIS grafts in in vivo models of esophageal repair.56,57
Conclusion
The results presented in this study detail the ability of bi-layer SF scaffolds to support attachment, proliferation, and differentiation of human GI epithelial and SMC lines in vitro. In comparison to conventional SIS matrices, bi-layer SF scaffolds promoted increased extents of attachment for each SMC line examined as well as a greater propensity for eEP cell differentiation toward suprabasal and superficial phenotypes. Future in vivo evaluations in models of defect repair are necessary to determine the potential of these scaffolds for GI organ reconstruction. In summary, bi-layer SF scaffolds represent promising platforms for GI tissue engineering.
Footnotes
Declaration of conflicting interest
The authors declare that there is no conflict of interests regarding the publication of this article.
Funding
The authors wish to thank the Office of Sponsored Programs at Boston Children’s Hospital for pilot study support as well as NIH/NIDDK T32-DK60442 and the Tissue Engineering Resource Center, NIH/NIBIB P41 EB002520.