
Abstract
Biomaterials that provide three-dimensional support networks for the culture of cells are being developed for a wide range of tissue engineering applications including the regeneration of bone. This study explores the potential of the versatile ionic-complementary peptide, FEFEFKFK, for such a purpose as this peptide spontaneously self-assembles into β-sheet-rich fibres that subsequently self-associate to form self-supporting hydrogels. Via simple live/dead cell assays, we demonstrated that 3 wt% hydrogels were optimal for the support of osteoblast cells. We went on to show that these cells are not only viable within the three-dimensional hydrogel but they also proliferate and produce osteogenic key proteins, that is, they behave like in vivo bone cells, over the 14-day period explored here. The gel elasticity increased over time when cells were present – in comparison to a decrease in control samples – indicating the deposition of matrix throughout the peptide scaffold. Moreover, significant quantities of calcium phosphate were deposited. Collectively, these data demonstrate that ionic-complementary octapeptides offer a suitable three-dimensional environment for osteoblastic cell function.
Introduction
The majority of work within cell biology over the past few decades has been carried out in vitro on two-dimensional (2D) systems, such as tissue culture plastic (TCP). 1 There has been a move more recently to develop and use three-dimensional (3D) scaffolds for the study of cells as these materials better reflect their in vivo environment and hence give more reliable results.2 –4 For example, the adhesion and migration behaviour of mammalian cancer cells displayed different behavioural patterns within 3D materials in comparison with cells cultured in 2D on flattened substrates.5 –7 Several classes of materials have been tested and used for the fabrication of 3D matrices for hosting and culturing cells, including polymers, protein and peptide-based materials. The latter is currently attracting significant attention as they inherently offer many of the required properties of a biomaterial, including immunocompatibility, biodegradability, easy handling, tunable structure and mechanical properties and ease of introducing functionality.3,4,8
These peptide-based hydrogels are typically fabricated from short amino acid sequences that spontaneously self-assemble into β-sheet- or α-helix-rich fibres. These go on to self-associate to form a self-supporting matrix that resembles natural extracellular matrix (ECM) when above a critical concentration.3,4,9 Consequently, many studies have focussed on the development of these scaffolds for the culture of several cell types, including bone-forming cells for bone tissue regeneration. For example, Zhang et al. developed the ionic-complementary peptide sequence, RADARADARADARADA (RADA-16), where R, A and D are arginine, alanine and aspartic acid, respectively. They showed that this sequence forms extended and ordered β-structured nanofibres that form self-supporting hydrogels. This system has since been used to support the 3D culture of mammalian cells including human osteoblasts (HOBs).10,11 Pochan and Schneider studied β-sheet hairpin peptides, such as VKVKVKVKVDPLPTKVKVKVKV-NH2 (MAX 1), where V is valine, K is lysine and P is proline. This amphiphilic peptide self-assembles into β-hairpin secondary structures in response to a change in environmental conditions, such as salt concentration. This leads to the formation of stable fibrous hydrogels capable of supporting the culture of various cells, such as mesenchymal stem cells, osteoblasts and murine fibroblasts.4,12,13 Beniash et al. 14 developed surfactant-based amphiphilic peptide hydrogels, which encapsulated and supported the proliferation of HOBs, making this system suitable for bone tissue engineering applications. One other β-sheet forming peptide – Ace-QQRFEWEFEQQ-NH2 (P-11) (Q, F, E and W are glutamine, phenylalanine, glutamic acid and tryptophan, respectively) – developed by Aggeli et al. 15 has been shown to facilitate teeth enamel re-mineralisation via hydroxyapatite nucleation.
It is widely known that some bone diseases such as osteoporosis and periodontal disease induce a progressive and significant loss of bone. Periodontitis, in particular, can cause the loss of teeth at late stages.16,17 Currently, deep scaling and root planning (debridement) is the standard method to treat this pathology. 18 Nonetheless, when a significant quantity of bone is lost, bone grafting is needed, but this unfortunately comes with several drawbacks, such as surgical complications, residual pain and increased risk of infection. In addition, some osteogenic factors are not completely viable after transplantation, and this has a negative effect on any bone regeneration. 19
Here, we wish to build on our groups’ previous work on the self-assembling behaviour and application of the peptide FEFEFKFK. In this octapeptide, F is non-charged and hydrophobic, and E and K are negatively and positively charged, respectively, at physiological pH. 20 FEFEFKFK self-assembles into an antiparallel β-sheet secondary structure, where all Fs are placed on the same side of the β-sheet while the charged amino acids are located on the other side. This peptide subsequently forms a network of β-sheet-rich nanofibres, which branch to form a self-supporting hydrogel depending on the pH, temperature and ionic strength of the media.20,21 Recently, we demonstrated that hydrogels prepared using FEFEFKFK were able to encapsulate bovine chondrocyte cells within 3D and the matrix supported their viability and proliferation. 21
Here, we extend this work and explore the suitability of our FEFEFKFK-based hydrogel to act as a scaffold for the 3D culture of primary HOB cells and the subsequent deposition of minerals key for the formation of bone. First, we explore the optimal mechanical strength of hydrogel required for cell viability. Second, we explore cell viability over longer culture times and proliferation, as well as the production of ECM proteins within the gel. Finally, we monitor the capability of the gel to facilitate the bone mineralisation process.
Materials and methods
Hydrogel preparation
FEFEFKFK peptide (>95% purity) was purchased from Biomatik Corporation, Canada. Peptide solutions were prepared initially at 2, 3, 4 or 5 wt% by dissolving the peptide powder in 800 µL of doubly distilled water (ddH2O) (see Table 1). They were subsequently vortexed and centrifuged (4000 r/min), before placing in an oven at 90°C for 2 h to ensure complete dissolution. To neutralise the sample, 1 M NaOH and Dulbecco’s phosphate-buffered saline (DPBS) were added, respectively (Table 1). This induced gelation. The resulting transparent gel was again vortexed and centrifuged (5000 r/min) and placed back into the oven at 90°C for 12–24 h before being cooled at room temperature (RT) to ensure formation of a homogeneous hydrogel.
FEFEFKFK gel concentrations and buffer conditions used for the preparation of hydrogels ready for the 3D culture of human osteoblast cells.

3D: three-dimensional; PBS: phosphate-buffered saline.
Oscillatory rheology
The viscoelastic behaviour of the FEFEFKFK hydrogels at days 0, 1, 3, 7 and 14 of cell culture was determined using an ARG2 rheometer with a 20-mm parallel plate geometry. Samples with and without cells were run in parallel, where the latter acted as the control. In each case, ~150 µL of gel was pipetted onto the bottom plate and left for 10 min to equilibrate before recording the elastic (G′) and viscous (G″) moduli as function strain (0.01%–100%) at 1 Hz and oscillatory frequency (0.1–100 Hz) at 1% strain. All samples were maintained at physiological temperature (37°C) using a Peltier stage and solvent evaporation minimised using a solvent trap.
Cell culture
HOBs (PromoCell, Heidelberg, Germany) were grown and maintained under standard cell culture conditions in Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% foetal bovine serum (FBS), 1% penicillin, 1% streptomycin and 50 µg/mL ascorbic acid. At 80%–90% of confluence, cells were subcultured using 0.05% trypsin–0.53 mM ethylenediaminetetraacetic acid (EDTA) 4Na solution (Gibco–Invitrogen, UK) and transferred to a Falcon tube to be centrifuged to remove the trypsin solution. The cell pellet was then resuspended in fresh medium and adjusted to the required cell concentration.
3D cell culture within FEFEFKFK hydrogel
Gels were sterilised by exposure to ultraviolet (UV) radiation for 30 min, and 200 µL of cell suspension was then pipetted on top and gently mixed by stirring with the pipette tip before gently pipetting up and down to create a homogeneous gel/cell suspension. A volume of 250 µL of gel containing 1.5 × 105 cells was transferred into each well in a 12-well cell culture insert (ThinCert™ Greiner Bio-One) for all experiments except rheology, where 500 µL of gel containing 2.5 × 105 cells was used. In addition, 1 × 104 cells/cm2 were pipetted directly onto a tissue culture plate. The gel was left to set for 10 min at 37°C. Gels were then placed in an incubator at 37°C in a 95% humidified atmosphere (20% O2) with 5% CO2 and fresh media changes were repeated five times at 20-min intervals to stabilise the pH at 7.2. Thereafter, the media were changed every 2 days to aid cell growth within the gel.
Cell viability
Cell viability was tested qualitatively using a live/dead assay (Invitrogen) and quantitatively using standard cell counting (haemocytometer). For the live/dead assay, 1.5 mL of PBS containing 2.5 µL of 4 µM ethidium homodimer-1 (EthD-1) assay solution and 1.5 µL of 2 µM calcein AM assay solution was prepared. The live/dead assay solution was pipetted on top of each hydrogel and then incubated under standard cell culture conditions for 20 min. The staining solution was then removed and samples were viewed under a Leica TCS SP5 confocal microscope. For quantitative cell viability, gels containing cells were rinse twice in PBS, diluted using trypsin (0.05%)–EDTA·4Na (0.53 mM). Subsequently, fresh cell culture media were added and mixed before transferring the cell suspension into a micro-centrifuge tube. Here, it was diluted 1:1 in trypan blue dye to enable counting the cells using a haemocytometer. Cell counts and optical images were obtained using an inverted microscope (Leica DM IL) and a SPOT insight camera (model 3.2.0; Diagnostic Instruments Inc., Michigan, USA).
Osteoblast proliferation
Osteoblast proliferation was assessed using PicoGreen® dsDNA assay (Life Technologies, Carslbad, CA, USA). After gels were rinsed twice with DPBS, they were resuspended in 500 µL of cold lysis buffer (200 mM Tris–HCl, 20 mM EDTA/ddH2O/1% Triton-X100) for 25 min. To ensure complete lysis, samples were vortexed vigorously and subject to three freeze–thaw cycles. Thereafter, samples were vortexed again and 100 µL gel–cell suspension was plated into black 96-well plates, where 100 µL of a working solution of Quant-iT PicoGreen reagent was added.
The samples were incubated for 2–5 min at RT. Fluorescence readings were obtained using a plate reader (FLUOstar OPTIMA; BMG LABTECH) at wavelengths of 435 nm (excitation) and 529 nm (emission). A standard curve was determined using calf thymus DNA in serial dilutions in 1% Triton X. A blank gel was used to correct the background absorbance and the assay was performed in triplicate.
Immunocytochemistry
The presence of collagen type I (col-I) inside the FEFEFKFK hydrogels was determined visually using a standard immunocytochemistry method after 7 and 14 days of culture. After 24 h of culture, each sample was prepared by culture in osteogenic media containing 10−5 mM dexamethasone (D4902-16; Sigma–Aldrich Co.), 10 mM β-glycerophosphate (G9422-100G; Sigma–Aldrich Co.) and 2.83 × 10−7 mM ascorbic acid (A8960-5G; Sigma-Aldrich Co., St. Louis, MO, USA). At each time-point, the gels were fixed with 3% paraformaldehyde (PFA) for 30 min at RT. Thereafter, samples were permeabilised using Triton X-100 at 0.05% in DPBS for 15 min at RT. Samples were subsequently blocked with 1% bovine serum albumin (BSA) for 40–50 min at RT and incubated for 1 h at RT with a primary antibody (pAb) (diluted in 1% BSA) rabbit polyclonal to col-I (Abcam, UK), at a ratio stipulated by the manufacturer (1:250). Subsequently, the samples were incubated for 1 h at RT in the dark with a goat anti-rabbit IgG-Alexa Fluor 594 (Abcam) as secondary antibody (sAb) along with Alexa fluor 488 phalloidin to target col-I and F-actin, respectively. The samples were rinsed in cycles of 5 × 5–10 min, with DPBS washings between each step. Samples were mounted on glass slides using ProLong Antifade Reagent (Invitrogen), and images were obtained using a Leica TCS SP5 confocal microscope.
Quantification of bone mineralisation proteins
Bone formation was monitored by quantifying mineralisation proteins that were present within each gel at days 7 and 14. At each time-point, gels were rinsed twice with DPBS and resuspended in cold distilled water to ensure gel dissolution. Cells within these samples were subjected to three freeze–thaw cycles which led to cell lysis. Three markers were selected and procedures are outlined below. In each case, a blank gel was used to correct the background absorbance.
Quantification of collagen
Collagen production was determined using a total collagen assay (QuickZyme Biosciences, Park, Leiden, The Netherlands). After resuspending and lysing, samples were vortexed and hydrolysed over 20 h at 95°C with 100 µL 12 M HCl. Samples were subsequently diluted 1:1 in 4 M HCl and pipetted into a 96-well plate, before adding 75 µL of an assay buffer and incubating for 20 min at RT. Following this, 75 µL of a detection reagent was added and the sample transferred into the oven (60°C) for 1 h. Absorbance measurements were obtained at 570 nm, using a plate reader (Tecan Infinite M200). Three independent assays were undertaken in triplicate.
Quantification of alkaline phosphatase activity
Alkaline phosphatase (alkphos) activity was monitored using a colorimetric alkphos and peroxidase substrate detection system (Sigma–Aldrich Co.). A volume of 20 µL of the cell lysis was added to transparent 96-well plates together with 200 µL of p-nitrophenyl phosphate (pNPP) solution (1 mg/mL pNPP, 0.2 M Tris buffer in 5 mL ddH2O) (SIGMAFAST™ pNPP Tablets, N1891-50SET; Sigma–Aldrich Co.), and the following reaction was stopped with 3 M NaOH. The absorbance of samples was measured using a plate reader (Labsystems Multiskan Ascent; Thermo Scientific, UK) at 405 nm every 30 s for 30 min. Two independent assays were performed in triplicate.
Quantification of osteocalcin
Osteocalcin (OCN) production was determined using a Human Osteocalcin ELISA Kit (Invitrogen–Life Technologies). After the cells were lysed, samples were vortexed and pipetted into a 96-well plate. Subsequently, a working anti-OST-HRP solution (100 µL) was added to each well, the plate covered and incubated for 2 h at RT. Each well was then rinsed three times with a washing solution before adding 100 µL of a chromogen solution (tetramethylbenzidine) and incubating for 30 min at RT in the dark. The reaction was stopped by adding 100 µL of a stop solution (1 N HCl). Absorbance measurements were obtained within 1 h at 450 nm, using a plate reader (Tecan Infinite M200). Two independent assays were performed in triplicate.
Mineralisation activity
Mineralisation activity was evaluated using Alizarin Red staining. At days 7 and 14, the cells were fixed with 70% ethanol for 30 min, rinsed three times with ddH2O and then stained with 40 nM Alizarin Red at RT in the dark for 45 min. Samples were then rinsed eight times with PBS. For optical density measurements, 10% cetylpyridinium chloride (CPC) was dissolved in 10 mM sodium phosphate and 1 mL of this was added to each gel before incubating at RT for 30 min. A volume of 200 µL of each solution was plated into a clear, flat-bottomed 96-well plate (Nunc, UK) and the absorbance at 562 nm determined using a Labsystems Multiskan Ascent plate reader. The assay was performed in triplicate.
Statistical analysis
Statistical significance in rheology, the PicoGreen, col-I, alkphos activity and OCN quantification assays, was determined using one-way analysis of variance (ANOVA) followed by post hoc comparisons (Tukey’s method).
Results and discussion
Initial experiments were undertaken with the aim of optimising the concentration of peptide hydrogel needed for the encapsulation, maintenance and proliferation of HOBs. It is known that varying the peptide concentration influences the mesh size of the network formed and also the mechanical strength of the hydrogel, both of which influence the flow of nutrients and waste products and also cell behaviour. To this end, four different concentrations of FEFEFKFK peptide hydrogel were prepared (2, 3, 4 and 5 wt%) in cell culture media and their elastic properties determined. Each concentration formed a stable, self-supporting hydrogel at pH 7 where the elastic modulus, G′, increased with increasing concentration: 6.2 ± 1.2, 7.8 ± 0.5, 13.3 ± 3.5 and 24.6 ± 3.5 kPa for the 2, 3, 4 and 5 wt% samples, respectively. This is in agreement with our previous work, and others, where the density of the fibre network increases with concentration.20 –22 The magnitude of the G′ values is higher than those obtained when the peptide was dissolved in pure water (in the order of 10–100 s Pa for the concentration range studied here). This is due to the salt within the cell culture media and PBS screening the charges of the peptide, leading to an enhancement of the hydrophobic forces and hydrogen bonding. This increases fibre aggregation and association, which increases the strength of the 3D network. 21
The effect of the changing mechanical properties on cell viability was subsequently explored by seeding each gel with 1.5 × 105 cells and monitoring cell fate via a live/dead assay over 7 days (Figure 1). When cells were incorporated within the 2 wt% sample, the resulting gels were rather weak and not stable, hence it was decided this system was not suitable for further study. It was clear that osteoblast viability was favoured when cells were cultured within 3 wt% gel where the majority of cells were alive (green) with only a few dead cells (red), in comparison to a majority of dead cells in both 4 and 5 wt% samples (Figure 1). It is postulated that this is due to the higher density of the fibre network which leads to smaller gel porosity, which perhaps is limiting the diffusion of nutrients, the exchange of gases and the output of cellular waste, or simply because the cells prefer to interact with the softer network. Consequently, 3 wt% gels were selected for further cell studies.
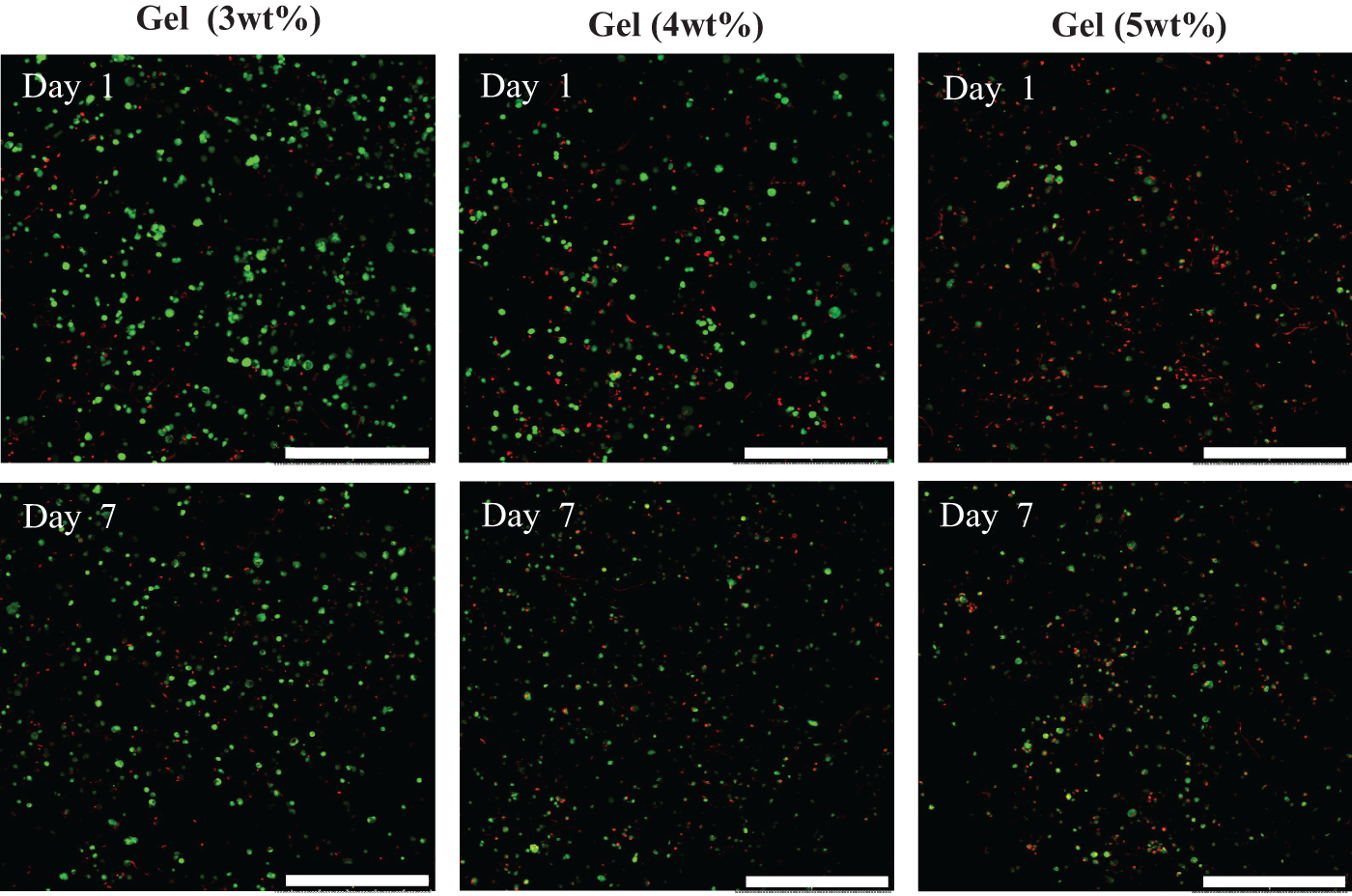
Live/dead imaging representing living cells (green) and dead cells (red) within FEFEFKFK hydrogel prepared at distinct peptides concentrations (magnification = 10×, scale bar = 500 µm).
Cell viability within 3 wt% FEFEFKFK gel samples was extended up to 14 days, and fluorescent micrographs of the live/dead assay and optical micrographs monitoring cell morphology over time are given in Figure 2. It is evident from live/dead screening that the majority of cells within each gel after 1, 7 and 14 days in culture were alive, with only a very few dead cells (Figure 2(a)–(c)). Importantly, cells were also incorporated throughout the peptide hydrogel, as evidenced by the even cell distribution in the Z-direction shown in the confocal image in Figure 2(d) (~250 µm thickness). Such entrapment and viability is similar to our previous work with bovine chondrocytes 21 and demonstrates the hydrogels can support cells within 3D. The optical micrographs in Figure 2(e)–(g) reveal that the HOBs typically acquired a rounded morphology during their 2 weeks in culture within the peptide hydrogel. When HOBs are present within natural bone, and their synthesis activity is induced, they tend to acquire a cuboidal/plump 3D morphology. This is in contrast to the rather flattened morphology commonly seen for bone lining cells, and also when the cells are grown on many 2D substrates.23,24 This suggests that the HOBs within our peptide hydrogel adopt an in vivo–like morphology, suggesting the peptide hydrogel is mimicking the 3D in vivo matrix environment. Such behaviour has been observed previously when HOBs have been cultured within synthetic poly(ethylene glycol) (PEG) 22 and natural alginate scaffolds. 25 It was also observed that HOBs formed some clusters throughout the FEFEFKFK hydrogel. These are also naturally present during osteoid synthesis. 24 To quantify the viability of HOBs within the peptide gels over their 2 weeks in culture, the percentage of viable cells was determined using the trypan blue exclusion quantification method. It is clear from these data presented in Figure 3 that the percentage viability of HOBs increases over the whole 14 days. This increase is most significant over the first 7 days.

(a-c) Live/dead imaging of HOB viability within 3 wt.% FEFEFKFK hydrogels during 14 days of culture. Living and dead cells are represented by green and red fluorescence respectively. Magnification=10×, scale bars 500 mm). (d) Axial (Z-axis) imaging show homogeneous HOB dispersion through the gel. (e-g) Optical micrographs (grey) show cell morphology within the gels. Magnification=10×, scale bars represent 100 mm.d. Axial (Z-axis) imaging show homogeneous HOB dispersion through the gel.

Quantification of cell viability as a function of culture time. Data are reported as mean values ± standard deviation for n = 3.
To identify the rate of proliferation of HOBs within the peptide hydrogel, the DNA content was quantified over time using a PicoGreen assay. It is clear from the data presented in Figure 4(a) that the DNA content within the gel increased significantly over the first 3 days of culture, and then remained steady, within experimental error, up to day 14. Such an increase in cell proliferation over the first few days has been seen for HOBs encapsulated within different types of hydrogels such as peptide amphiphile 14 and alginate gels. 25 A decrease in DNA content over longer time periods has also been reported for HOBs when cultured within, for example, PEG hydrogels. These possess higher mechanical properties (10–300 kPa) in comparison to the FEFEFKFK hydrogel studied here.22,26 It is known that calcified bone possesses high stiffness,22,27 which is superior to the strength of a typical hydrogel system,21,22,25 therefore there is a possibility that the relatively low mechanical strength provided by our peptide hydrogel might influence the rate of proliferation of HOBs (i.e. decrease) over prolonged periods of time. This is unlikely in this case, however, given the cells were not viable within the stiffer 4 and 5 wt% gels. Furthermore, gels are dynamic systems and as such are prone to expose less surface area to cells over time. This might also impact on the extent and rate of cell proliferation. The DNA content quantified for HOBs cultured in 2D on TCP is given in Figure 4(b). A similar pattern is evident here; a significant increase over the first few days which then remains constant over longer times. This behaviour is typical for 2D cell culture and is likely to be due to over-confluence at later times (Figure 4(b)).

Quantification of DNA content from HOBs cultured (a) in 3D within FEFEFKFK peptide hydrogel and (b) in 2D on TCP. Data are reported as mean ± standard deviation for n = 3.
It is also important to consider that HOBs are specialised cells, and as such, their predominant role is the secretion of proteins. It is believed that their activation and proliferation are triggered mainly by systemic factors such as hormones and other local factors, which include cytokines and growth factors released after bone resorption occurs. The role of osteocytes sensing micro-alterations or mechanical loads on bone tissue is also very important to activate HOBs. Finally, the autocrine activation via bone-specific ECM proteins is fundamental to induce the proliferation of osteoblasts.24,28,29 The rate of proliferation of HOBs within 3D culture systems is, therefore, complex and clearly dependent on cell culture conditions used, such as type of scaffold, type of cells and/or the cell densities used. Here, we demonstrated that our system is able to support the viability and proliferation of HOBs, without any additional cell proliferative factor. Consequently, the next step is to test the ability of cells to produce key proteins for bone formation, including collagen-I (col-I). Figure 5 shows the fluorescence and immunocytochemistry images of stained gels for the identification of F-actin that defines the cell architecture, and col-I, at days 7 and 14. It is clear that there is significant production of col-I inside the cell cytoplasm over the first 7 days of culture. There is also some evidence of extracellular col-I staining, albeit with less intensity. After 14 days in culture, however, a high concentration of col-I was detected, not only inside the cells but also surrounding the cells, particularly in areas of the gel where cells formed clusters. The high concentration of col-I suggests that cells are functional within the gel and consequently synthesise proteins to remodel their niche. This confirms that HOBs can survive in the in vitro hydrogel and moreover produce ECM proteins that are important for their survival and bone formation.

Fluorescence micrographs of actin cytoskeleton staining (green) and collagen I production (red) by HOBs cultured in 3D within 3 wt% FEFEFKFK hydrogel over 14 days (magnification = 10×, scale bar = 500 µm).
To corroborate the visual trend observed for the production of col-I from the immunocytochemistry staining experiments, the quantity of protein present was determined (Figure 6(a)) using a quantitative colorimetric method. Total collagen was found to increase between days 7 and 14, which fits well with qualitative results observed in Figure 5. Such an increase in collagen production suggests that the cells might also be producing other ECM proteins involved in the process of bone formation, such as alkphos and OCN. These have, therefore, also been quantified and data reveal a similar pattern as collagen (Figure 6(b) and (c), respectively). The data for the production of all three ECM proteins for the 2D culture of cells on TCP cultures were also determined and showed similar increasing production over time, as expected. Such ECM protein production within the FEFEFKFK hydrogel is a significant finding, as it highlights it as a promising scaffold for the culture of bone-forming cells. It also demonstrates the gels’ potential for the culture of other types of mammalian cells. The pattern of ECM production exhibited by the HOBs is similar to that observed by other groups using different types of scaffolds, including poly(

Total collagen production within (a) gel and (b) TCP; alkaline phosphatase activity within (c) gel and (d) TCP and osteocalcin production inside (e) gel and (f) TCP over 14 days in culture. Data are reported as mean ± standard deviation for n = 3 (col-I) and n = 2 (alkphos and OCN).
It is known that cell responses can be influenced by the characteristics of the niche, including stiffness, topography and roughness. 27 The natural ECM matrix that hosts bone in vivo has a high content of non-mineralised collagen and has a compressive modulus stiffness of ~100 kPa, which increases to ~1000 kPa once osteoid mineralises.22,27 Both these values are significantly higher than the elastic modulus reported for peptide hydrogels.11,14,21 Nevertheless, we are not aiming to match the in vivo mechanical strength in our FEFEFKFK hydrogel, instead we are exploring our gels’ ability to host HOBs and the ability of the cells in this environment to synthesise and release in situ bone-forming proteins, and consequently deposit calcium phosphate, for non-load bearing applications. The effect of HOBs and the production of matrix protein on the stiffness of the peptide hydrogel was monitored using oscillatory rheology. As a control, we also studied the stiffness of a gel without cells that had identical media changes over time. The results are given in Figure 7 and indicate that the elasticity of the gel network without cells decreased from 8 to 3.2 ± 0.6 kPa over 14 days in culture media. This drop is likely due to the natural dissolution of gel that inevitably occurs during media changes, given our peptide hydrogel is a physical gel. In contrast, the elasticity of the FEFEFKFK hydrogel with cells increased from ~5.4 to 22.6 ± 1.2 kPa over the 14 days of culture. This differing behaviour is most likely to be due to the cells remodelling the gel and the deposition of ECM proteins. A similar trend has been reported by other authors for FEFEFKFK hydrogel 21 and also the commercial PuraMatrix 32 systems for the culture of chondrocytes.
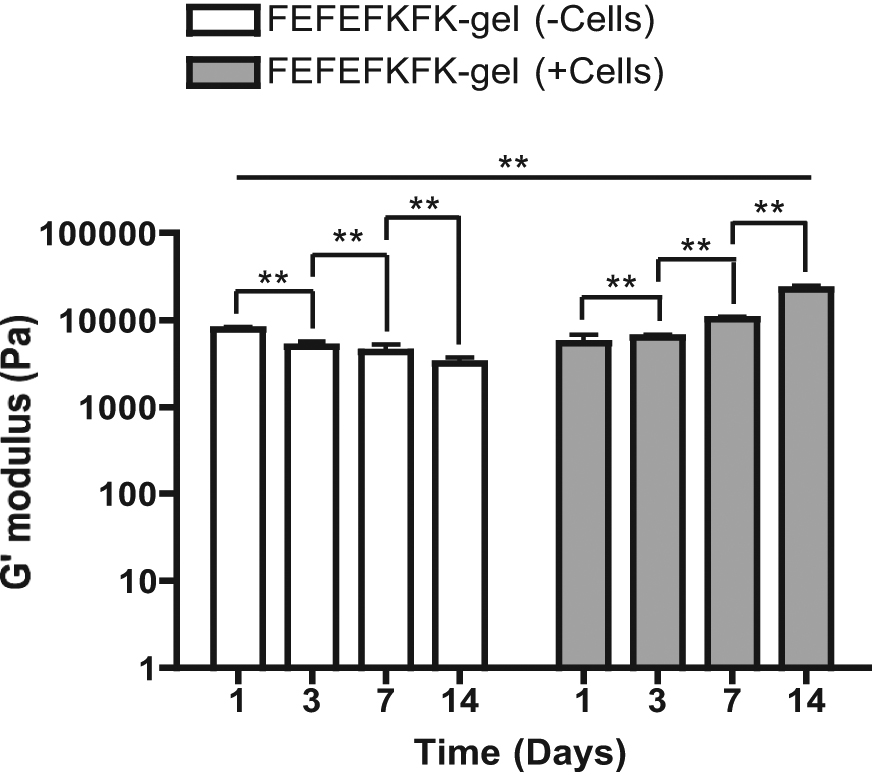
Elastic modulus, G′, of FEFEFKFK hydrogels over 14 days in cell culture media conditions, both with and without the presence of HOBs. Data are reported as mean ± standard deviation for n = 3.
Bone mineralisation is the last phase in the bone formation process, where several proteins such as OCN, osteopontin and bone sialoprotein are expressed. 33 It is known that during this process, osteoblasts release vesicles containing minerals such as calcium and phosphate, which are essential for healthy bone formation.23,33,34 The increasing presence of OCN during culture within the hydrogels has been monitored and the results are shown in Figure 6. We went on to monitor the presence of any calcium deposits using Alizarin Red staining, both qualitatively from imaging and quantitatively using absorbance at 562 nm. The photographs of the stained samples are shown in Figure 8(a) where the intensity of the red stain indicates the presence of calcium and consequently confirms calcium phosphate formation. It is clear that discrete calcium deposits are present within the gels after the first 7 days of culture, with even stronger staining intensity observed after 14 days (Figure 8(a)). Staining of 2D cell culture on TCP was also observed, but the signal was not as intense as in the 3D studies. This was expected given there is only a thin film present for the 2D experiments. Quantitative results are given in Figure 8(b) where the absorbance of the specific dye was recorded at 562 nm over time. It is clear that in both 3D and 2D cases, the mineralisation assay confirms a significant increase in mineralisation over time within the gels (Figure 8(b) and (c), respectively). The intensity of absorbance is an order of magnitude higher for the sample where cells were incorporated throughout the hydrogel. This indicates that the FEFEFKFK hydrogel not only supports, maintains and allows the proliferation of primary HOBs but also allows the production of ECM proteins and the deposition of calcium phosphate and hence shows bone formation. Other work on different systems report similar increases in the deposition of calcium over time that ranges from 7 to 21 days in culture.22,30,35,36

(a) Alizarin red staining images showing the presence of calcium phosphate deposits in red both in FEFEFKFK hydrogel, and on 2D culture (TCP). Quantification assay of calcium deposits within (b) gel and (c) TCP, using Alizarin Red staining via extraction with 10% CPC. Data are reported as mean ± standard deviation for n = 3.
Conclusion
Here, we have demonstrated that a 3 wt% FEFEFKFK fibrillar hydrogel is an excellent host for the 3D culture of primary HOB cells. Cell viability and proliferation are maintained over 14 days within the gel (maximum length of time tested), as is the production of bone matrix proteins. Mineralisation within the hydrogel has been shown and correlates nicely with an increase in the mechanical properties of the gel over the 14 days in culture. These ionic-complementary peptide gels have potential for use as 3D bone regeneration scaffolds and as in vitro osteoblast culture systems.
Footnotes
Acknowledgements
The authors would like to thank the members of the Polymers and Peptides, and Biomaterials and Tissue Engineering groups for helpful discussions.
Declaration of conflicting interest
The authors declare that there is no conflict of interest.
Funding
LCD acknowledges a PhD scholarship grant from CONACyT, Mexico