
Abstract
The development of a biosimilar is based on comparative structural, physicochemical, functional and clinical assessments. The sum of these analyses encompasses the ‘totality of evidence’, which demonstrates no clinically meaningful differences between the biosimilar and the reference product (RP). Once biosimilarity has been established, provided there is suitable scientific justification, clinical data may be extrapolated to other indications of the RP. AVT02 has been developed as a biosimilar to high-concentration, low-volume Humira (adalimumab), an anti-tumour necrosis factor-alpha monoclonal antibody approved for various chronic inflammatory indications. The totality of evidence for AVT02 is described, supporting its approval as an adalimumab biosimilar for all approved indications globally. Analytical similarity assessments using mass spectrometry methods demonstrated identical amino acid sequences for AVT02 and the RP, with high similarity in terms of primary structure, post-translational modifications and higher-order structural attributes. The mechanism of action was assessed by various cell-based potency assays and binding assays, and the results demonstrated that AVT02 is highly similar to the RP. No clinically meaningful differences in terms of purity, potency and safety were observed, and minor differences in a few physiochemical attributes did not impact the in vitro biologic activity and were not considered clinically relevant. Clinical similarity was demonstrated by comparing the pharmacokinetic, efficacy, safety and immunogenicity profiles of AVT02 with those of the RP. Clinical studies supported similar pharmacokinetic and comparable immunogenicity profiles between AVT02 and the RP in healthy participants and participants with moderate-to-severe chronic plaque psoriasis, with no new safety signals detected. The totality of evidence described demonstrates the biosimilarity of AVT02 to the RP, thereby fulfilling the scientific and regulatory requirements for AVT02 as a high-concentration biosimilar for the treatment of chronic plaque psoriasis and all approved indications of the RP.
Plain language summary
Biosimilars are drugs that have similar quality, effectiveness, and safety profiles to an already approved biological drug, which is referred to as the ‘reference product (RP)’. Although biosimilars have identical amino acids (the building blocks that make up proteins) to the RPs, they are manufactured in living cells which leads to a small amount of natural variability. Therefore, extensive testing is required to confirm that a biosimilar is highly similar to the RP. The ‘totality of evidence’ is a set of tests to demonstrate that there are no meaningful differences between the biosimilar and the RP, in other words, that there is ‘biosimilarity’ between the biosimilar and RP. Once biosimilarity has been proven, the biosimilar may be used to treat all the diseases currently treated with the RP, without the need for separate clinical trials in each disease. AVT02 has been developed as a biosimilar to Humira, an antibody approved for various chronic inflammatory diseases such as chronic plaque psoriasis (PsO). A step-by-step approach was used to show biosimilarity of AVT02 to Humira. This included clinical studies (in healthy individuals and participants with moderate to severe chronic PsO) and non-clinical studies (comparisons of the chemistry of the drugs and how they work in the body). Clinical studies in healthy individuals and participants with PsO showed that AVT02 and Humira were taken up and degraded by the body in a similar way, peoples’ immune response to the two drugs were similar, and both drugs had similar side effects. No clinically meaningful differences in the purity, effectiveness, and safety of AVT02 compared with Humira were seen. The evidence demonstrates the biosimilarity of AVT02 to Humira and supports the use of AVT02 to treat all conditions which are currently treated with Humira.
Keywords
Introduction
Biosimilars are biologic drugs that have similar quality, efficacy and safety profiles to an already approved reference product (RP). 1 Biosimilars share an identical amino acid sequence but have inherent variability as they are manufactured using living cells. Since biosimilars do not have the full ‘sameness’, extensive analytical testing is required to confirm that they are highly similar to the RP. 2 The development of a biosimilar is based on thorough comparative analytical assessments using a comprehensive panel of analytical methods that are comprised of structural and functional assays, and comparative clinical pharmacokinetics (PK) and/or pharmacodynamics (PD), as well as comparative clinical efficacy and safety (including immunogenicity) assessments. The sum of these analyses encompasses the ‘totality of evidence’ supporting a conclusion of biosimilarity.3,4 Application of established knowledge and experience with the RP, in terms of safety, quality and efficacy combined with an appropriate clinical program, is adequate to confirm the similarity of the biosimilar to the RP. 4
Once biosimilarity has been established, provided there is suitable scientific justification, clinical data may be extrapolated to other indications of the RP. 5 According to relevant European Medicines Agency (EMA) and US Food and Drug Administration (FDA) guidance, an applicant may seek approval of a proposed biosimilar for one or more additional indications for which the RP is approved, via extrapolation of clinical efficacy and safety data not specifically studied during the clinical development of the proposed biosimilar and based on the overall evidence of similarity provided from the similarity exercise.3,5 The scientific justification can be based on a combination of knowledge of the mechanism(s) of action, PK, PD, efficacy, safety and immunogenicity of the RP in each of its approved indications. 6
AVT02 has been developed as a biosimilar to the high-concentration, low-volume versions of its RP Humira® (adalimumab, ATC code L04AB04). 7 RP adalimumab is an anti-tumour necrosis factor-alpha (TNF-α) monoclonal antibody globally approved for various indications, including chronic plaque psoriasis (PsO), rheumatoid arthritis (RA), psoriatic arthritis (PsA), inflammatory bowel diseases (IBDs) and ankylosing spondylitis (AS). 7 Although high and low concentrations of RP are available, the majority of RP prescriptions are for high concentration preparation in most countries. 8 AVT02 is marketed as a high-concentration, low-volume (100 mg/ml), citrate-free preparation. Adalimumab formulations with high-concentration and citrate-free offer advantages over the 50 mg/ml preparation, as its higher-concentration and lower-volume preparation makes it more patient-friendly with less injection site-related pain.9–12 The route of administration, dosing regimen and dosage form of AVT02 are the same as approved for the RP. The clinical similarity of AVT02 to the RP was previously assessed in two comparative studies: a PK study in healthy participants and a confirmatory efficacy and safety study in participants with chronic PsO, which supported the biosimilarity of AVT02.10–12 This article describes the totality of evidence demonstrating biosimilarity of AVT02 to the RP, and the scientific justification for the extrapolation across approved disease indications for the RP.
The high cost of RP adalimumab may preclude some patients from being able to access the treatment, especially considering prolonged use is required to treat the chronic conditions indicated for this product. A similar product that provides comparable safety and efficacy at reduced cost would fulfil a broader medical need as a more cost-effective treatment.13–15
Structure of adalimumab
Adalimumab (total molecular weight ~148 kDa) is a tetramer composed of two light kappa chains (~24 kDa each) and two heavy immunoglobulin G1 chains (~49 kDa each), each of the latter containing one N-linked glycosylation (glycan) site. 16 This molecule comprised 1330 amino acids with each light chain consisting of 214 amino acid residues and each heavy chain consisting of 451 amino acid residues. 17
Mechanism of action of adalimumab
The expression of TNF cytokine is tightly controlled, with elevated levels found in the affected tissues of patients with RA, juvenile rheumatoid arthritis (JIA), AS, PsA, PsO, ulcerative colitis and Crohn’s disease (CD), and is postulated to be involved in the pathophysiology of hidradenitis suppurativa (HS) and uveitis (UV).18–24 The anti-inflammatory effect of adalimumab can be mediated in multiple ways (Figure 1). 25

Overview of known and purported mechanisms of action for adalimumab: (a) sTNF-adalimumab trimer complexes, (b) reverse signalling, (c) CDC and (d) ADCC.
Adalimumab binds to both soluble [sTNF; Figure 1(a)] and transmembrane [mTNF; Figure 1(b)] TNF via the antigen-binding fragment (Fab) domain, blocking interaction with, and downstream signalling of, TNF receptor 1 (TNFR1) and TNFR2.26–28 The formation of sTNF-adalimumab trimer complexes disrupts receptor binding thus inhibiting the inflammatory cascade, leading to downregulation of adhesion molecules responsible for leukocyte migration (endothelial cell leukocyte adhesion molecule [ELAM]-1, vascular cell adhesion molecule [VCAM]-1 and intracellular adhesion molecule [ICAM]-1).29,30
In a TNF-producing cell, binding of adalimumab Fab region to mTNF may also, via cross-linking, induce ‘reverse signalling’, resulting in cytokine suppression and apoptosis [Figure 1(b)]. 31 Mechanisms involving the crystallizable fraction (Fc) region of adalimumab have also been proposed. Cytotoxic effects of adalimumab may be mediated through the interaction of the Fc region of mTNF-bound adalimumab with complement component 1q (C1q), leading to complement-dependent cytotoxicity [CDC; Figure 1(c)], and Fc receptors on effector cells leading to antibody-dependent cell-mediated cytotoxicity [ADCC; Figure 1(d)]. 31
The precise contribution of each mechanism of action per indication is not fully known.
Stepwise approach to establishing the biosimilarity of AVT02 to Humira
To support the demonstration of biosimilarity, a stepwise development approach was used based on EMA 5 and FDA 3 guidance documents and considering scientific advice from the agencies obtained at multiple time points during development. The approach comprised a comprehensive three-way comparative analytical similarity assessment of structure, purity and physiochemical and functional (potency and binding assays) properties of AVT02, US-Humira (US-RP) and EU-Humira (EU-RP) to support the demonstration that AVT02, US-RP and EU-RP (representative of global supply) are highly similar. A single-dose PK study in healthy individuals (Study AVT02-GL-101; NCT03849313) provided a three-way comparison of AVT02, US-RP and EU-RP intended to (i) support PK similarity of AVT02 and US-RP and EU-RP and (ii) provide a PK bridge to support the relevance of the clinical comparative data generated using EU-RP, to support a demonstration of the biosimilarity of AVT02 to US-RP. 11 A comparative clinical study between AVT02 and EU-RP in participants with moderate-to-severe chronic PsO (Study AVT02-GL-301; NCT03849404) was also conducted to support a demonstration of no clinically meaningful differences in terms of efficacy and safety (including immunogenicity). 10
Structural and functional assessments
Several head-to-head comparative analytical similarity assessments were conducted during the development of AVT02 and included analysis as part of Quality Target Product Profile assessments.
The extensive, comparative analytical similarity assessment of AVT02, EU-RP and US-RP using orthogonal analytical methods provided a comprehensive understanding and comparison of the respective amino acid sequences, molecular masses and post-translational modifications. The complementary mass spectrometry data from peptide mapping and analysis of intact and reduced mass, complemented by amino acid sequencing by Edman degradation, indicate that the amino acid sequences for AVT02, EU-RP and US-RP are identical (Table 1). AVT02 is also highly similar to the RP in terms of primary structure, post-translational modifications and higher-order structural attributes. Assessment of glycan by 2-aminobenzamide labelling and normal-phase high-performance liquid chromatography demonstrated overall good correlation between the glycan profiles of AVT02 and the RP. Some differences in the levels of specific glycans were observed that were non-meaningful. 32
Comparative analytical similarity assessment.
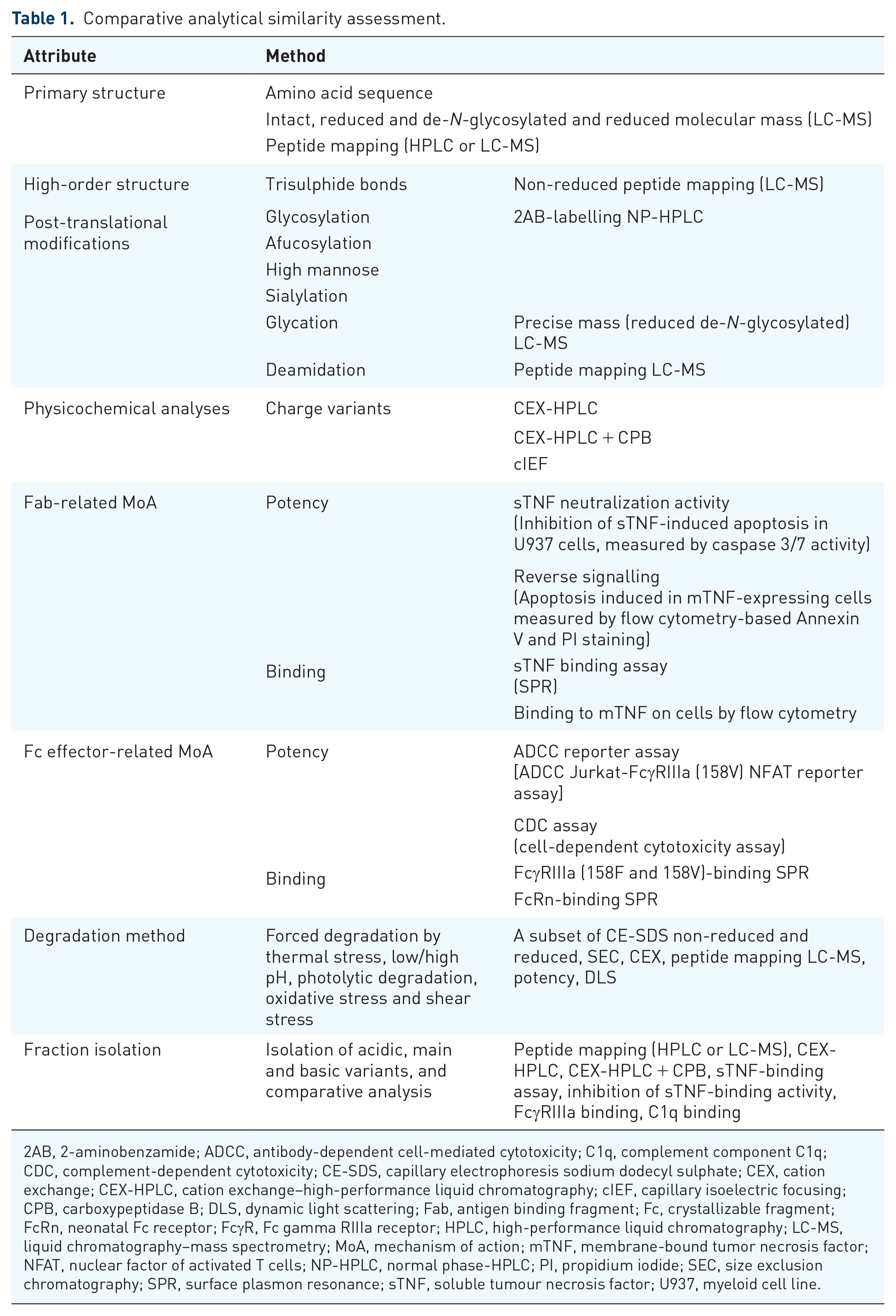
2AB, 2-aminobenzamide; ADCC, antibody-dependent cell-mediated cytotoxicity; C1q, complement component C1q; CDC, complement-dependent cytotoxicity; CE-SDS, capillary electrophoresis sodium dodecyl sulphate; CEX, cation exchange; CEX-HPLC, cation exchange–high-performance liquid chromatography; cIEF, capillary isoelectric focusing; CPB, carboxypeptidase B; DLS, dynamic light scattering; Fab, antigen binding fragment; Fc, crystallizable fragment; FcRn, neonatal Fc receptor; FcγR, Fc gamma RIIIa receptor; HPLC, high-performance liquid chromatography; LC-MS, liquid chromatography–mass spectrometry; MoA, mechanism of action; mTNF, membrane-bound tumor necrosis factor; NFAT, nuclear factor of activated T cells; NP-HPLC, normal phase-HPLC; PI, propidium iodide; SEC, size exclusion chromatography; SPR, surface plasmon resonance; sTNF, soluble tumour necrosis factor; U937, myeloid cell line.
Total afucosylation (including high mannose) for AVT02 was lower compared with the RP. This was due to lower levels of high-mannose glycans. This difference could potentially cause an impact on ADCC activity (afucosylation) and PK (high mannose); however, there was no effect on ADCC activity, as demonstrated by multiple orthogonal assays, and binding to FcγRIIIa or FcRn binding (key PK indicator) was not impacted. In addition, study AVT02-GL-101 demonstrated PK similarity of AVT02 and the RP; therefore, the observed differences in high mannose do not impact PK similarity. 11 Additionally, the forced degradation study revealed no new product-related impurities identified in AVT02 that were not present in the RP, indicating that AVT02 has the same primary degradation routes as the RP.
Deamidation, oxidation and N- and C-terminal integrity were assessed by liquid chromatography–mass spectrometry peptide mapping. Levels of deamidation were low overall and were highly similar to the majority of deamidation sites. Minor differences observed in deamidation, oxidation and C-terminal heterogeneity did not result in differences in the binding and biological activity data and were considered not to be clinically meaningful or relevant. Based on these comparative analytical similarity assessments, AVT02 is highly similar to these attributes in EU-RP and US-RP, and EU-RP is highly similar to US-RP.
Further characterization using physicochemical analyses confirmed that AVT02 has a comparable absorption coefficient to the RP and the same charge-variant characteristics. Charge variants were analysed by cation exchange–high-performance liquid chromatography and capillary isoelectric focusing. The distribution of charge variants differed between AVT02 and the RP, mainly due to the C-terminal lysine clipping of the heavy chain. These differences did not lead to any detectable decrease in biological activity or function.
The functional characterization of AVT02 included an extensive collection of orthogonal and sensitive potency and binding assays. These were designed to examine both the Fab region functionalities of the molecule and the Fc-mediated functionalities of adalimumab, thus providing a complete functional comparison of AVT02 and the RP. Furthermore, a series of bioassays were specifically performed to support the extrapolation of AVT02 function to different clinical indications of the RP. These bioassays are particularly informative when examining the different hypothesized mechanisms of action (MoAs) for adalimumab and comparing AVT02 with the RP.
The primary MoA of adalimumab was evaluated with orthogonal potency assays. AVT02 inhibition of TNF-induced apoptosis was demonstrated to be highly similar to the RP in the sTNF-neutralization potency assay.
The inhibition of apoptosis was also evaluated in human intestinal epithelial cells, further demonstrating a highly similar response for AVT02 compared to the RP and allowing for extrapolation to gut-associated indications. The hypothesized MoA for the RP, reverse mTNF-induced signalling and apoptosis were shown to be highly similar to AVT02 with regard to efficacy in CD.
These functional cell-based assays were supported by sensitive binding assays to both sTNF and mTNF. For sTNF, a surface plasmon resonance (SPR) assay showed highly similar binding for AVT02 and the RP. In the case of mTNF, the cell-based assay demonstrated a highly similar response for AVT02 and the RP. Binding to FcRn was evaluated using an SPR-based method and revealed AVT02 to be highly similar to the RP. This result was further confirmed by the clinical study AVT02-GL-101 which demonstrated clinical PK equivalence between AVT02 and the RP. 11
Data from the ADCC peripheral blood mononuclear cell assay, supported by data from the reporter assay, showed a high similarity between AVT02 and the RP. This finding was strengthened by SPR binding studies to Fc receptors (FcγRIIIa, FcRn, FcγRIa, FcγRIIa, FcγRIIb, FcγRIIIb), which showed highly comparable binding between AVT02 for all FcγRs as well as FcRn. Other than ADCC, it is also important to evaluate the potential MoA involving the use of the immune complement system to mediate cell lysis of mTNF-expressing cells, to which adalimumab is bound. This CDC cell-based assay demonstrated AVT02 to be highly similar in potency to the RP.
Finally, the induction of regulatory macrophages and inhibition of CD4+ T cells by AVT02 and the RP were compared in a two-way mixed lymphocyte reaction using two different donor pairs, with both groups demonstrating a highly similar response. This mechanism is not only Fab dependent but also shown to be Fc dependent and is a hypothesized MoA for the treatment of CD and IBD with the RP. 33
Based on the totality of data from the comprehensive and analytical similarity assessments, AVT02 is considered to be highly structurally and functionally similar to the RP.
Clinical similarity assessments
Clinical studies were conducted to confirm the similarity of AVT02 and the RP, established from the structural and functional assessments (Table 1). The clinical development program was designed to meet the requirements outlined in the FDA and EMA guidelines for biosimilars.3,5 These clinical studies demonstrated the clinical similarity of AVT02 to the RP by assessing PK profiles of AVT02, EU-RP and US-RP, 11 as well as the therapeutic equivalence of AVT02 to EU-RP. 10 The PsO population was selected as the most sensitive population for detecting any potential differences between AVT02 and the RP since it has the greatest placebo-adjusted response rate (61–64%). 34 Indications with the highest placebo-adjusted response rate are considered the most sensitive for detecting any potential difference between the proposed biosimilar and the RP (data on file). PsO is also the most sensitive indication for detecting any potential difference in safety and immunogenicity since patients are usually treated with biologic monotherapy, without the potential adverse events and impact on the immunogenicity of concomitant medication (such as methotrexate)35,36 commonly used in other adalimumab-approved indications. The decision to use the most sensitive indication to detect change increases the likelihood of detecting any small difference between the test product and RP and is in line with regulatory expectations for the clinical development of biosimilars.
Clinical PK
The AVT02-GL-101 study evaluated the three-way PK similarity, safety and immunogenicity of AVT02 compared with EU-RP and US-RP. An innovative adaptive study design was used to optimize sample size even with the limited availability of information on the variability of PK parameters for the new high-concentration formulation (100 mg/ml) of adalimumab. A two-stage adaptive design was used to minimize the risk of underpowering/overpowering the study, making it one of the first examples of adaptive study design in a biosimilar setting.
In this PK study, three pairwise comparisons (AVT02 to EU-RP, AVT02 to US-RP and EU-RP to US-RP) in 390 adult male and female healthy subjects receiving a single subcutaneous (s.c.) dose of 40 mg of AVT02, EU-RP or US-RP exhibited highly similar PK profiles (Figure 2). 11 All pairwise comparisons met the pre-specified acceptance criteria for PK similarity [90% confidence intervals for the ratios of geometric mean of Cmax, area under the serum concentration–time curve from time zero (pre-dose) to the time of the last quantifiable concentration (AUC0–t) and area under the serum concentration–time curve from time zero (pre-dose) extrapolated to infinity (AUC0–inf) within the interval of 80–125%] [Figure 3(a) and (b)]. PK similarity of AVT02 to both EU-RP and US-RP was demonstrated. 11

Overview of PK similarity assessment of adalimumab primary PK (AVT02-GL-101, PK population). 11

Mean serum concentration–time profiles of adalimumab by treatment group on (a) linear and (b) semi-logarithmic scales (AVT02-GL-101, PK population). 11
Further support for the PK similarity of AVT02 and EU-RP was gained from the AVT02-GL-301 study, which revealed no clinically meaningful difference in the PK parameter and trough concentrations (Ctrough) between the proposed biosimilar AVT02 and the RP in individuals with moderate-to-severe chronic PsO. 10 Ctrough values of AVT02 and EU-RP in this study were detectable at week 4 and remained stable up to the final measurement. Mean Ctrough levels of AVT02 and EU-RP were comparable at all time points.
Based on the comparative PK data, combined with knowledge of the PK profiles of the RP in different patient populations, 37 the PK profile and biodistribution of AVT02 are expected to be similar to EU-RP and US-RP (based on analytical and PK bridging data) in all approved indications.
Clinical efficacy
The AVT02-GL-301 study was a confirmatory efficacy and safety trial that compared the efficacy profiles of AVT02 and the RP in the most sensitive indication – participants with moderate-to-severe chronic PsO. 10 This was a 54-week, randomized, double-blind, parallel-group study conducted with 412 adult participants. Participants were randomized 1:1 to AVT02 or EU-RP at a dose of 80 mg (2 × 40 mg) subcutaneously in week 1, then 40 mg every other week. At week 16, participants treated with EU-RP were re-randomized 1:1 to undergo a single transition to AVT02 or to continue to being treated with EU-RP up to week 48. The primary efficacy endpoint was the percent improvement in the Psoriasis Area Severity Index (PASI) score from baseline to week 16. The robustness of these efficacy results was confirmed by the following sensitivity analyses: analysis of covariance (ANCOVA), including site as a random effect; ANCOVA of week 16 completers in the full analysis set (FAS) and mixed model for repeated measures analysis in the FAS and per protocol set for the percent improvement in PASI score from baseline to week 16. The percent improvement in PASI score at week 16 was similar in both groups: 89.2% in the AVT02 group and 86.9% in the RP group [Figure 4(a)]. The ANCOVA of percent improvement from baseline in PASI score demonstrated that AVT02 is well within the predefined equivalence margins of ±10% for the 90% confidence interval at week 16 in the FAS, defined as all randomized participants who received at least one dose of randomized study drug. The percent improvement from baseline in PASI score at weeks 4, 8, 12, 16 and 50 showed very similar levels of improvement in participants randomized to AVT02 and the RP in the last observation carry-forward analysis [Figure 4(a) and (b)].

LS mean (±SE) of percent improvement from baseline in PASI score by visit – FAS through (a) week 16 and (b) week 50 (AVT02-GL-301). 10
Analysis of other secondary (e.g. static Physician’s Global Assessment) and exploratory efficacy (e.g. Routine Assessment of Patient Index Data 3) parameters as well as subgroup analyses of efficacy [e.g. in anti-drug antibody (ADA)-positive and -negative participants or neutralizing antibody (nAb)-positive and -negative participants] through week 16 and week 50 (final study-related efficacy assessment) showed no clinically meaningful differences between the AVT02 and EU-RP treatment groups.
AVT02 has demonstrated similar clinical efficacy to EU-RP in the most sensitive population (moderate-to-severe chronic PsO). 10 Similar efficacy of AVT02 to the RP is expected in all approved indications.
Clinical safety
Overall, safety data from five clinical trials of AVT02 with a product representative of the commercial manufacturing process are available: two single-dose studies in healthy individuals [AVT02-GL-101 and AVT02-GL-102 (NCT03983876)]11,12; two studies in individuals with moderate-to-severe chronic PsO [AVT02-GL-301 and AVT02-GL-302 (NCT04453137)] and one study in individuals with moderate-to-severe active RA [AVT02-GL-303 (NCT04224194)].10,38,39 Comparative safety data between AVT02 and EU-RP were collected in individuals with PsO in the AVT02-GL-301 comparative clinical efficacy and safety study. 10 No clinically meaningful differences in the safety profiles of AVT02, EU-RP and US-RP, including events of special interest, were observed following single s.c. administration of 40 mg adalimumab to healthy individuals. 11 Additionally, in both studies (AVT02-GL-101 and AVT02-GL-301), the frequency of injection site reactions and low injection site pain rates were similar to the RP, supporting the attributes of the new high-concentration citrate-free formulation.10,11
The safety results obtained in the non-comparative clinical trials (AVT02-GL-303 and AVT02-GL-102) were in line with the known safety profile of the RP and did not reveal any unexpected signals compared with what has previously been observed in the clinical development of AVT02. Overall, results from the five clinical studies demonstrate that the safety profile of AVT02 is consistent with information reported in the US Prescribing Information and EU Summary of Product Characteristics of the RP.7,40 Therefore, the AVT02 clinical development program safety results support a demonstration of biosimilarity, as no clinically meaningful differences were observed, and results were consistent with the well-characterized safety profile of the RP. Therefore, similar safety profiles of AVT02 to the RP are anticipated across all approved indications.
Clinical immunogenicity
Biologics such as adalimumab are known to elicit immune responses, causing the body to produce ADAs and nAbs that could affect PK and pharmacodynamic profiles, reduce the efficacy and bioavailability of the drug, cause adverse effects or result in treatment failure.41,42 The immunogenicity profiles (ADA and nAb) of AVT02 and RP were similar in both comparative clinical studies AVT02-GL-101 and AVT02-GL-301 (Tables 2 and 3). The incidence of immunogenicity in the PK similarity study (AVT02-GL-101) was high, 11 as expected in healthy participants and well known for the RP, and similar between treatment arms (AVT02/EU-RP/US-RP). The immunogenicity profiles (ADA and nAb) of AVT02, EU-RP and US-RP were comparable in healthy individuals following single-dose administration. 11 At the end of the study (day 64), the frequency of ADAs and nAbs was comparable for AVT02, EU-RP and US-RP, and positive detection of binding ADAs was observed in >95% of subjects (Table 2). At day 64, the frequency of nAb-positive subjects was comparable across the three treatment groups: 80.6% in the AVT02 group, 86.9% in the EU-RP group and 87.0% in the US-RP group. 11
Frequency count (%) of ADAs and nAbs to adalimumab over time (AVT02-GL-101). 11

ADA, anti-drug antibody; EoS, end of study; N, number of treated patients; nAb, neutralizing antibodies; RP, reference product.
Frequency count n (%) of ADAs and nAbs to adalimumab over time in PASI responders (AVT02-GL-301) (data on file).
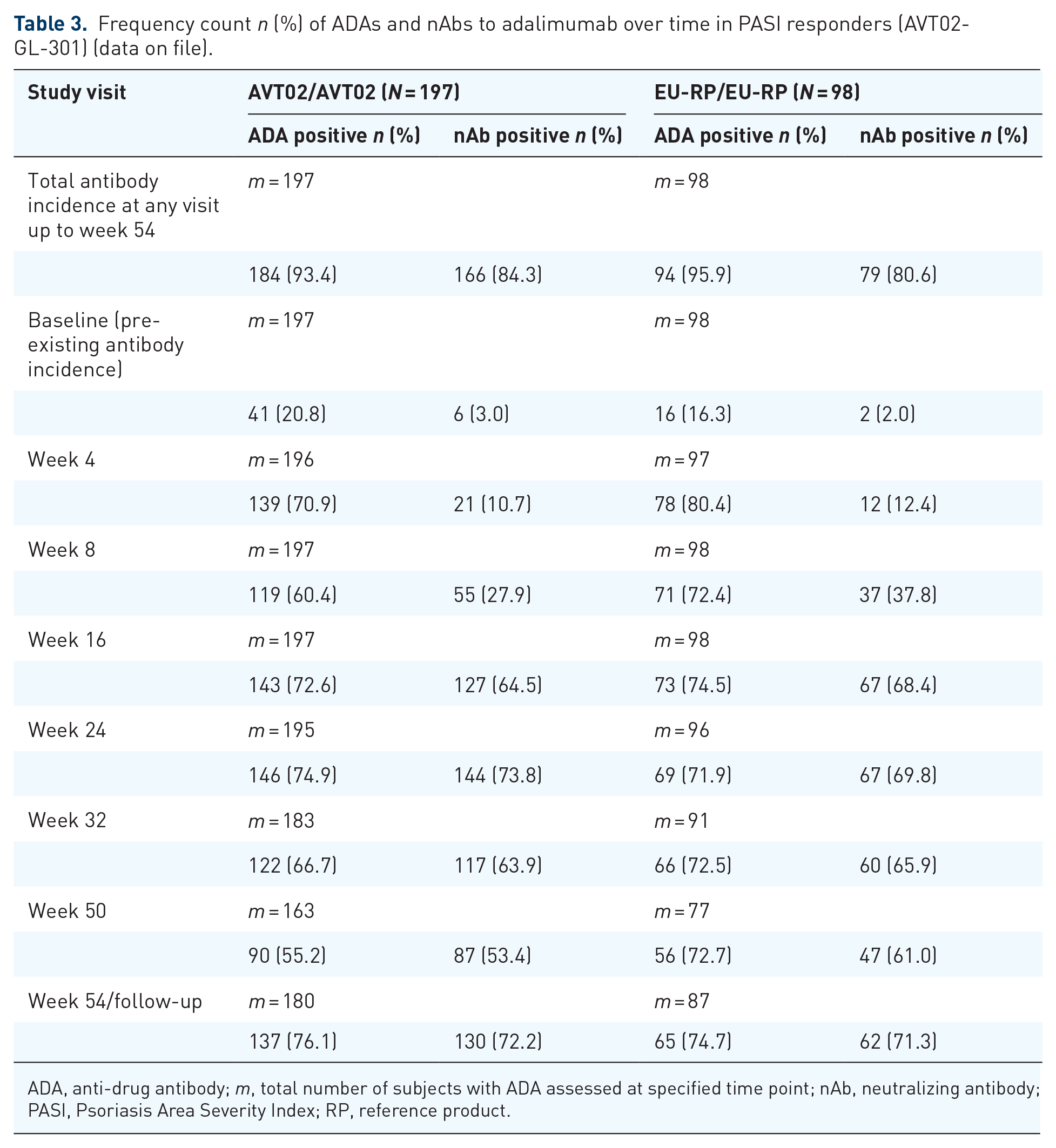
ADA, anti-drug antibody; m, total number of subjects with ADA assessed at specified time point; nAb, neutralizing antibody; PASI, Psoriasis Area Severity Index; RP, reference product.
Immunogenicity in the comparative PsO clinical trial AVT02-GL-301 in subjects with moderate-to-severe chronic PsO was similar in individuals treated with AVT02 and the RP. 10 ADAs and nAbs were detected in most participants during the observation period irrespective of treatment. 10 The majority of participants (>93%) tested positive for ADAs over the 54-week period (Table 3). Of these participants, most also tested positive for nAbs over the same period. There was no difference in adalimumab serum trough concentrations when comparing participants treated with AVT02 or EU-RP in subgroups with or without ADAs, or with nAbs throughout the entire study period. 10
The immunogenicity results contribute to the totality of the evidence, thus supporting the demonstration of biosimilarity between AVT02 and the RP. Similar immunogenicity profiles of AVT02 to the RP are expected in all approved indications.7,40
Extrapolation of indications
Extrapolation of data and information from direct studies of a biosimilar in a particular indication can be used to scientifically justify approval for other indications. Scientific justification can be based on a combination of factors, including knowledge of the mechanism(s) of action, PK, PD, efficacy, safety and immunogenicity of the RP in each of its approved indications. 6 If no differences which could affect these factors are identified between the biosimilar and the RP, approval of the biosimilar in additional indications, not studied specifically, may be supported.
Based on the totality of data from the comprehensive and comparative analytical similarity assessment, AVT02 was shown to be highly structurally and functionally similar to the RP.10,11 Binding of AVT02 to TNF (associated with inflammatory cascade and reverse signalling), 31 FcγRIIIa (associated with ADCC) and C1q (associated with CDC) 31 were shown to be comparable between AVT02 and RP adalimumab. While the precise contribution of each of these modes of action per indication for adalimumab is not fully elucidated, the physicochemical characterization, as well as cell-based functional assays, show that AVT02 can be expected to be biosimilar to RP adalimumab in all MoA scenarios, thereby supporting extrapolation to all indications.
The guidelines call for an ‘adequately sensitive’ or ‘appropriately sensitive’ population in which to detect potential differences between a candidate biosimilar and the RP.3,5 PK similarity was demonstrated in a single-dose study in healthy volunteers, 11 considered a sufficiently sensitive and homogeneous population that is least likely to have PK variability. Comparable efficacy was demonstrated in the PsO indication, 10 selected as the most sensitive population for detecting any potential differences between AVT02 and the RP since it has the greatest placebo-adjusted treatment effect of all approved indications in the RP. Results showing comparable efficacy between AVT02 and RP adalimumab in PsO support extrapolation to the other, less sensitive, indications. PsO was also selected as the most sensitive indication in which to detect potential differences in safety and immunogenicity profile, since the patient population in the AVT02 clinical development program was prescribed adalimumab as monotherapy, without potential biasing factors that would decrease sensitivity, for example, combination therapy with methotrexate, a typical approach in RA.
Together, the similarity of critical quality attributes of AVT02 compared with RP adalimumab, leading to the comparability of the mode of action(s), as well as the clinical similarity in an ‘adequately’ and ‘appropriately’ sensitive population of AVT02 compared with RP adalimumab, along with the well-understood immunogenicity and safety profile across indications in all licensed indications of the RP comprise the scientific justification for and support an application of, the safe and efficacious use of AVT02 across all approved indications of the RP.
Conclusion
The development of AVT02 as a biosimilar to the RP has produced a body of analytical, functional and clinical data that assess the comparative similarity between AVT02 and the RP, which encompasses the totality of the evidence. No clinically meaningful differences in terms of purity, potency and safety were observed, and minor differences did not impact the in vitro biological activity and were not considered clinically meaningful. Clinical studies supported similar PK and comparable immunogenicity profiles between AVT02 and US-RP and EU-RP in healthy individuals and participants with moderate-to-severe chronic PsO receiving AVT02 and EU-RP (similar to US-RP based on analytical and PK bridging data), with no new safety signals detected. The totality of evidence described demonstrates the biosimilarity of AVT02 to the RP, thereby fulfilling the scientific and regulatory requirements for high-concentration formulation of AVT02 as a biosimilar. Therefore, in addition to biosimilarity and similar analytical, PK, safety and immunogenicity profiles, it is expected that AVT02 would have a similar benefit–risk profile as the RP across all approved indications.
Supplemental Material
sj-pptx-1-taj-10.1177_20406223231223286 – Supplemental material for The totality of evidence approach in the development of AVT02 (adalimumab), a biosimilar to Humira
Supplemental material, sj-pptx-1-taj-10.1177_20406223231223286 for The totality of evidence approach in the development of AVT02 (adalimumab), a biosimilar to Humira by Joseph E. McClellan, Sesselja Ómarsdóttir, Nivedita Roy, Verena Berger, Cecilia Michel and Fausto Berti in Therapeutic Advances in Chronic Disease
Footnotes
Acknowledgements
Correction (March 2024):
Article updated with the addition of online supplemental material.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.