
Abstract
Adrenocortical carcinoma (ACC) is a rare, aggressive malignancy with an annual incidence of ~1 case per million population. Differentiating between ACC and benign adrenocortical tumors can be challenging in patients who present with an incidentally discovered adrenal mass, due to the limited specificity of standard diagnostic imaging. Recently, urine steroid metabolite profiling has been prospectively validated as a novel diagnostic tool for the detection of malignancy with improved accuracy over current modalities. Surgery represents the only curative treatment for ACC, although local recurrence and metastases are common, even after a margin-negative resection is performed. Unlike other intra-abdominal cancers, the role of minimally invasive surgery and lymphadenectomy in ACC is controversial. Adjuvant therapy with the adrenolytic drug mitotane is used to reduce the risk of recurrence after surgery, although evidence supporting its efficacy is limited; it is also currently unclear whether all patients or a subset with the highest risk of recurrence should receive this treatment. Large-scale pan-genomic studies have yielded insights into the pathogenesis of ACC and have defined distinct molecular signatures associated with clinical outcomes that may be used to improve prognostication. For patients with advanced ACC, palliative combination chemotherapy with mitotane is the current standard of care; however, this is associated with poor response rates (RR). Knowledge from molecular profiling studies has been used to guide the development of novel targeted therapies; however, these have shown limited efficacy in early phase trials. As a result, there is an urgent unmet need for more effective therapies for patients with this devastating disease.
Introduction
Adrenocortical carcinoma (ACC) is a rare endocrine malignancy that arises from the cortex of the adrenal gland and has an estimated worldwide incidence of ~1 case per million population per year.1 –3 Despite its rarity, ACC generally portends a poor prognosis as the majority of patients develop locally recurrent or metastatic disease, despite undergoing seemingly curative surgical resection. 4 A number of questions and controversies exist regarding aspects of diagnosis, medical treatment, and the surgical management of ACC. The rarity of this disease, and short duration of survival, has resulted in a paucity of prospective clinical trials. Therefore, current treatment recommendations are largely driven by consensus opinion based on retrospective data.5,6 While our understanding of the molecular mechanisms that underpin ACC development has greatly improved as a result of large-scale pan-genomic studies,7,8 the translation of this knowledge into effective clinical therapies for patients with advanced disease has been limited to date. In this review, we provide an overview of the current state of the art in the diagnosis and treatment of ACC, highlighting ongoing controversies and recent advances, as well as their potential effect on clinical practice.
Epidemiology
ACC is a rare malignancy that meets the criteria for ‘orphan’ disease designation in the European Union and in the United States (<50 cases per 100,000 population and <64 cases per 10,000 population, respectively). 9 Analysis of the National Cancer Institute’s Surveillance, Epidemiology and End Results (SEER) database indicates an annual incidence of ACC of 0.72–1.02 per million in the United States.1,2 This is in accordance with a population-based study from the Netherlands, which reported an incidence of 1 case per million. 3 In contrast, the incidence of ACC is 10-fold higher in Southern Brazilian children, which is partly attributable to the high frequency of a specific founder tumor protein 53 (TP53) germline mutation in this population. 10 Although ACC can occur at any age, some studies have described a bimodal age distribution with an initial peak in incidence during childhood, followed by another in the fourth to fifth decade of life.11,12 Others have depicted only a single peak with a median age at diagnosis of 55–56 years.1 –3 Despite this, the observation that ACCs account for 1.3% of all pediatric cancers 13 versus ~0.02% of malignant tumors in adults,14,15 suggests a relatively higher incidence in childhood. Regardless of age, ACC shows a slight female predilection (ratio of affected females to males: 1.5–2.5:1).11,16 Epidemiological risk factors for ACC are not well understood, although recent studies have identified an increased incidence in male cigarette smokers.17 –19 In females, estrogen exposure has been implicated as a risk factor due to an increased incidence of ACC in users of the oral contraceptive pill, 19 and the anti-proliferative effects of estrogen inhibition on ACC cells in vitro. 20
Pathogenesis
Initial insights into the genes and signaling pathways involved in ACC tumorigenesis came from studies of familial diseases associated with ACC development, for which the causative germline alterations are well-defined. These include: Li-Fraumeni syndrome (caused by inactivating TP53 mutations); 21 Beckwith-Wiedemann syndrome [caused by genetic and epigenetic abnormalities of chromosome 11 (11p15.5)]; 22 multiple endocrine neoplasia (MEN) type 1 (caused by inactivating MEN1 mutations); 23 and Lynch syndrome [caused by inactivating mutations in the mismatch repair genes MutL homolog 1 (MLH1), MutS homolog 2 (MSH2), MutS homolog 6 (MSH6), and PSM1 homolog 1, mismatched repair system component (PMS2)]. 24 However, the vast majority of ACCs are sporadic; it is increasingly understood that they may be driven by a myriad of genetic and epigenetic aberrations. These include somatic DNA mutations, chromosomal aneuploidy, altered DNA methylation, and dysregulated microRNA (miRNA) expression. 25
Much of our current understanding of the landscape of molecular alterations in ACC is derived from two tour de force studies: one from the European Network for the Study of Adrenal Tumors (ENS@T) 7 and another from The Cancer Genome Atlas Study on ACC group (ACC-TCGA). 8 Both consortia utilized a range of multi-omics techniques, including DNA exome sequencing, mRNA expression profiling, miRNA profiling, DNA methylation analysis, and single nucleotide polymorphism (SNP) arrays that enabled in-depth pan-molecular characterization of a large number of primary ACCs for the first time. Although a detailed discussion of these findings is beyond the scope of this review and has been provided recently by others,25 –27 both studies showed that ACCs harbor somatic driver mutations that most frequently affect genes involved in Wnt/β-catenin signaling [Zinc and ring finger 3 (ZNRF3), Catenin Beta 1 (CTNNB1)]; cell cycle regulation [TP53, cyclin dependent kinase inhibitor 2A (CDKN2A), retinoblastoma protein 1 (RB1), cyclin-dependent kinase (CDK)]; and chromatin remodeling [MEN1, death domain associated protein (DAXX)].7,8 They also confirmed findings from earlier single platform studies28,29 which showed that overexpression of the insulin-like growth factor-2 (IGF2) occurs in 80–90% of ACCs, largely due to loss of heterozygosity at the IGF2 locus at chromosome 11p15 (i.e. loss of the imprinted maternal allele and duplication of the paternal allele).7,8 Importantly, these studies have enabled the classification of ACC into molecular subtypes with distinct biological signatures associated with good and poor prognosis, and have identified potentially druggable targets.
Clinical features
ACC generally presents in three forms: ~40–60% of patients present with symptoms and signs of hormone excess, ~30% present with nonspecific symptoms (e.g. abdominal pain and fullness due to tumor growth or constitutional symptoms of malignancy), and 20–30% are asymptomatic and diagnosed incidentally on cross-sectional imaging performed for other indications. 30 The most common syndrome of hormone excess in patients with functional ACC is Cushing’s syndrome, which occurs in ~45% of patients and may cause central weight gain, plethora, proximal muscle atrophy, diabetes mellitus, and easy bruising. A mixed clinical picture of Cushing’s and virilization due to concomitant cortisol and androgen hypersecretion may also be seen in 20–30% of patients.31,32 The latter phenotype is strongly suggestive of malignancy and is not seen in patients with benign adrenal adenomas. 30 While autonomous aldosterone secretion is rare in ACC, hypokalemia and hypertension may still occur secondary to glucocorticoid-induced mineralocorticoid receptor activation in patients with hypercortisolism. 30 Feminization in males due to excess estrogen production occurs in 1–5% of patients. 33 The effect of functional status on survival outcomes in patients with ACC is controversial. Although some series have shown hormone secretion to be an independent risk factor for poorer overall survival (OS) and recurrence-free survival (RFS),34 –37 these findings have not been consistently confirmed by others.38,39 A meta-analysis by Vanbrabant et al. 40 demonstrated an increased risk of mortality [relative risk (RR) 1.71] and recurrence (RR 1.43) in cortisol-secreting, but not androgen-secreting, ACC. However, it is unclear whether these findings are due to the deleterious effects of cortisol hypersecretion rather than true underlying differences in tumor biology and aggressiveness.
Diagnostic evaluation
Clinical workup of suspected ACC is centered around hormonal evaluation and cross-sectional imaging, with a final diagnosis confirmed by histopathological examination. The combined rarity of this malignancy and the lack of disease-specific symptoms can make ruling out a diagnosis of ACC challenging in a patient with an incidentally discovered adrenal mass, (an adrenal ‘incidentaloma’) particularly when imaging and biochemistry are inconclusive.
Biochemistry
Biochemical evidence of hormone excess is found in 60–70% of patients with ACC.41,42 Hormonal evaluation is useful in the workup of ACC for several reasons: (1) the presence of steroid hormone excess establishes the adrenocortical origin of the tumor and obviates the need for unnecessary investigations such as biopsies; (2) the specific pattern of hormones excess (e.g. co-secretion of cortisol and androgens) may further raise suspicion for malignancy; (3) the measurement of hormone levels may serve as a useful biomarker for recurrence during follow-up after surgery; (4) the need for postoperative hydrocortisone replacement in patients with cortisol-producing ACCs can be determined; and (5) the potential for intraoperative hypertensive crises due to undiagnosed pheochromocytoma is avoided.5,30 Specific biochemical tests, as recommended in current ENS@T/European Society of Endocrinology (ESE) guidelines, 5 are shown in Table 1.
Hormonal and biochemical workup in patients with suspected ACC.

Table adapted from Fassnacht et al. 5
Can be omitted if hypercortisolism is excluded.
The most suitable set of precursors and sex hormones has not yet been established and local testing availability may dictate what tests are available.
ACTH, adrenocorticotropic hormone; DHEA-S, dehydroepiandrosterone sulfate.
Imaging
Computed tomography
Imaging forms the cornerstone of evaluation in patients with suspected ACC; the most commonly used first-line modality is typically an abdominal computed tomography (CT) scan. 43 CT features suggestive of an ACC rather than a benign adrenal mass include the presence of irregular borders, areas of necrosis, hemorrhage, and/or calcification [Figure 1(a)]. Invasion into surrounding structures, such as the inferior vena cava, may also be seen [Figure 1(b)]. 44 The risk of malignancy in an adrenal mass increases with size, with ACCs typically presenting with a median diameter of 10 cm. 31 The risk of ACC by size has been reported as 2%, 6%, and 25% for adrenal lesions of <4 cm, 4–6 cm, and >6 cm, respectively, leading to the recommendation in some consensus guidelines that adrenalectomy should be considered for lesions >4 cm.45,46 Calculation of tissue density as an approximation of intracellular lipid content on unenhanced CT, by measuring Hounsfield units (HU), is also used to distinguish between ACC and benign adrenocortical adenomas. Lipid-rich adenomas typically exhibit low attenuation on unenhanced CT and a HU threshold of ⩽10 is suggested as being indicative of a benign lesion in current ENS@T/ESE and European Society for Medical Oncology (ESMO) guidelines.33,47 In contrast, a HU of >10 is suggested as being indicative of malignancy. However, the distinction between benign and malignant adrenocortical tumors based on imaging is not always clear-cut, and the challenges surrounding this are discussed in detail elsewhere.33,48

CT imaging in a patient with advanced ACC. (a) Axial CT image showing a heterogeneously enhancing right adrenal mass (red arrow) measuring 9.9 × 6.0 × 8.0 cm with possible invasion into the liver. (b) Coronal CT image from the same patient showing tumor thrombus within the inferior vena cava (yellow arrow).
Positron emission tomography-computed tomography
The utility of [ 18 F]-fluorodeoxyglucose–positron emission tomography (FDG-PET)/CT in the evaluation of ACC is not well defined. In patients with a history of extra-adrenal malignancy, this modality may be useful in distinguishing between benign lesions and metastases originating from cancers that have a propensity for spreading to the adrenals (e.g., liver, lung, lymph nodes, and bone). 49 However, in patients without known or suspected extra-adrenal malignancy and indeterminate CT findings, the role of FDG-PET/CT is less clear. In a study of 87 patients without known cancer, who underwent workup for adrenal lesions with indeterminate findings on washout CT, Guerin et al. 50 demonstrated that an FDG adrenal lesion/liver standardized uptake value (SUV) ratio of >1.5 detected malignant adrenal lesions with reasonable sensitivity and specificity (86.7% and 86.1%, respectively). In contrast, in a study of 106 ACC patients, FDG-PET/CT only provided additional information beyond contrast-enhanced CT in a minority (5%) of patients. 51 Other studies evaluating FDG-PET/CT for the assessment of adrenal masses are limited by the inclusion of small numbers of ACC patients and suboptimal reporting of test accuracy; 48 therefore, there is a lack of consensus regarding whether FDG-PET/CT regarding should be recommended for routine use in all patients with suspected ACC. 5 Furthermore, limitations of this modality include cost, additional radiation exposure, and the potential for false-positive findings, particularly in functional adenomas and nonmetastatic pheochromocytomas, which may exhibit FDG-avidity. 49
Metomidate (MTO) is a potent inhibitor of the adrenal enzymes CYP11B1 (11β-hydroxylase) and CYP11B2 (aldosterone synthase). Given its selectivity for these adrenal-specific targets, MTO labelled with [ 11 C] for PET imaging or [ 123 I] for single-photon emission computerized tomography (SPECT)/CT has been developed as an alternative tracer for functional adrenal imaging. 52 Evidence to date suggests that while MTO may be useful in distinguishing between cortical and noncortical adrenal lesions, its ability to differentiate benign from malignant adrenal tumors is limited. 52 Despite this, a subset of patients with metastatic ACC have been shown to demonstrate [ 123 I] MTO uptake, which has provided the rationale for trials of radionuclide-based systematic therapy in this subgroup (discussed below).53,54
Urine steroid metabolomics
Limitations in the diagnostic accuracy of the imaging tests described above are reflected in the low prevalence of malignancy (<10%) in contemporary series of patients undergoing adrenalectomy for non-functioning incidentalomas.55,56 Therefore, patients are commonly subjected to multiple radiological studies and unnecessary surgical resection of adrenal masses that are ultimately revealed to be benign. A technology that has recently come to the forefront as a means of improving current standard-of-care workup for ACC is urine steroid metabolomics, which utilizes mass spectrometry-based urinary steroid metabolite profiling in combination with machine-learning-based data analysis. 42 The rationale for this approach is based on the finding that ACCs are relatively inefficient steroid producers, due to the dysregulated expression of steroidogenic enzymes. 57 This results in the excessive secretion of a range of steroid hormone precursors, instead of the normal end products of steroid hormone biosynthesis. These precursors, while not routinely measured in blood tests, can be detected using gas or liquid chromatography/mass spectrometry analysis of 24-hour urine samples. Proof-of-concept for this approach was first demonstrated by Arlt et al., 41 who retrospectively compared 45 patients with ACC to 102 patients with benign adrenal adenomas, showing that the former had significantly greater urinary excretion of androgen precursor metabolites, metabolites of active androgens, deoxycorticosterone, and glucocorticoid precursors. Using a machine-learning-based algorithm, a malignant steroid ‘fingerprint’ was generated, which predicted ACC with 90% sensitivity and 90% specificity. 41
The recently published Evaluation of Urine Steroid Metabolomics in the Differential Diagnosis of Adrenocortical Tumours (EURINE-ACT) study prospectively validated the panel proposed by Arlt et al. 41 in an international cohort of 2017 patients, 98 of whom had ACC. 58 In this study, three tests (tumor diameter, imaging characteristics, and urine steroid metabolomics) were assessed for diagnostic accuracy, either separately or in combination. The combination of all three tests demonstrated the highest positive predictive value (76.5%) for diagnosing ACC in patients with the following results: tumor size >4 cm, CT attenuation >20 HU, and a high-risk urine steroid metabolomics profile. The negative predictive value of this triple testing strategy was high, (99.7%) highlighting its potential value in ruling out ACC and preventing a subset of patients from undergoing unnecessary investigations and surgery. The same group has also shown, in a preliminary study, that urine steroid metabolomics can be used as for the detection of ACC recurrence after surgery; however, prospective validation and comparison against the reference standard for recurrence detection (imaging) is awaited. 59
Despite the clear benefits of a non-invasive assay such as urine steroid metabolomics, the widespread implementation of this technology into routine clinical practice is currently limited by both the cost and availability of mass spectrometry equipment as well as the need for inter-laboratory cross-validation of assay results.
Pathology
While the clinical, biochemical, and radiological tests outlined above may raise suspicion for ACC, the final diagnosis is made on histological examination, which should be performed by an experienced endocrine pathologist. Macroscopically, ACCs tend to be large lobulated masses [Figure 2(a)], with heterogenous areas of hemorrhage and necrosis within a fibrous capsule. Their cut surface ranges from brown to orange to yellow depending upon intracellular lipid content [Figure 2(b)]. The only definitive criteria for malignancy are the presence of distant metastasis and/or loco-regional invasion; in the absence of these features, the diagnosis may be made using various histological multiparameter scoring systems. In practice, the system first proposed by Weiss in 1984 60 is most widely used. The Weiss score is based on nine microscopic criteria, each of which is weighted equally and given a score of 1 if present (Table 2). 60 A total score of ⩾3 is consistent with ACC, while a score of 0–2 indicates an adrenocortical adenoma. Some borderline lesions scoring 2, however, are still be considered suspicious and may be deemed as having ‘uncertain malignant potential’. 61 The reliability of the Weiss system has been challenged in these borderline cases and, in recent years, additional scoring systems have been developed. These are either simplified versions of the existing Weiss score 62 and/or incorporate immunohistochemical staining for reticulin 63 or Ki-67. 64 Each of these demonstrates similarly high performance, with sensitivities of ~100% and specificities of 90–99% for diagnosing ACC. 65 Nonetheless, the very existence of several competing scoring systems highlights the challenges and complexities faced in the pathological assessment of ACC and the fact that no individual system is ideal. In an effort to standardize the reporting process, the International Collaboration on Cancer Reporting (ICCR) have proposed a minimum reporting standard of 23 items that should be included in ACC pathology reports, based on the consensus of an international panel of expert adrenal pathologists. 66

Gross pathology of a resected right-sided ACC. (a) Gross image showing a 10 cm brown mass with an attached rim of liver tissue that was resected en bloc. (b) Cut surface of the resected ACC showing a variegated appearance with focal areas of hemorrhage.
Weiss criteria. 60

A total score of >3 is suggestive of ACC.
uclear grading is based on the Fuhrman nuclear grading system used in renal cell carcinoma. 67
HPF, high power fields.
It should be noted that the Weiss score overdiagnoses malignancy in the oncocytic subtype of adrenocortical tumors. This is because oncocytic tumors inherently possess at least three features of the Weiss criteria (high nuclear polymorphism, <25% clear cells, and diffuse architecture), whether they are benign or malignant. 61 For this variant, a modified scoring system (the Lin-Weiss-Bisceglia system) 68 should be utilized. In contrast, the Weiss score underestimates the risk of malignancy in the even rarer myxoid variant of adrenal tumors. Although no specific scoring system exists for this subtype, the Helsinki score (which incorporates Ki-67) has been shown to outperform the Weiss score in this context. 65
Is there a role for preoperative adrenal biopsy?
Transcutaneous adrenal biopsy is not generally indicated in patients with ACC because the limited amount of tissue obtained from this procedure makes the differentiation between benign and malignant adrenal masses difficult. 69 Furthermore, complications may occur in 11% of patients, including hemorrhage, pneumothorax, pancreatitis, and the small risk of needle-track seeding due to violation of the tumor capsule. 70 Despite this, biopsy may be indicated in patients with metastatic ACC who are not surgical candidates and in whom a tissue diagnosis would help inform oncological management. Biopsy may also be useful in situations where the exclusion of suspected metastasis in patients with a known extra-adrenal primary malignancy would guide the choice of therapy (e.g. surgery for limited disease versus chemotherapy for metastatic disease). 5 Prior to biopsy being performed, biochemical exclusion of pheochromocytoma is necessary to avoid the possibility of potentially life-threatening catecholamine surge. 5
Staging and prognosis
Staging systems
Accurate staging of ACC plays a key role in treatment planning and prognostication. Although several staging systems have been described, the one proposed by the ENS@T group in 2009 is most widely used. 71 The ENS@T staging system is a modification of the original 2004 American Joint Committee on Cancer (AJCC)/Union for International Cancer Control (UICC) tumor, node, metastasis (TNM) classification (Table 3), which was criticized for its inability to discriminate between the survival outcomes of patients with stage II and III disease. 71 Furthermore, it was realized that patients with tumor invasion into surrounding adipose tissue and positive lymph nodes, or those with invasion into adjacent organs fared significantly better than those with distant metastases, despite all of these patients being classified as stage IV disease according to the 2004 AJCC/UICC system.71,72 Instead, the ENS@T system defines stage I and stage II ACC as tumors confined to the adrenal gland, that measure ⩽5 cm and >5 cm, respectively; stage III denotes tumors that extend into surrounding adipose tissue or adjacent organs or involve locoregional lymph nodes; stage IV only includes tumors with established distant metastases. 71 The changes proposed by ENS@T were recently incorporated into the 2017 (8th edition) AJCC/UICC staging manual for ACC, 73 such that both staging systems are now virtually identical (Table 3). Other groups have attempted to further refine the discriminatory ability of the ENS@T staging system with the inclusion of additional clinicopathological factors such as age,74,75 Ki-67,75,76 lymphovascular invasion, 77 resection margin status, 75 number of tumor-involved organs, 75 and the presence of symptoms at diagnosis. 75 Of these factors, Ki-67 appears to be the single most powerful predictor of disease recurrence following resection, with the longest RFS seen in patients with a Ki-67 index of <10%. 78
Comparison of UICC/AJCC 2004 and ENS@T 2009/UICC/AJCC 2017 staging classifications for ACC.

Tumors are classified as follows: T1, ⩽5 cm; T2, >5 cm tumor; T3, tumor infiltration into surrounding tissue; T4, tumor invasion into adjacent organs; N0, no positive lymph nodes; N1, positive lymph node(s); M0, no distant metastases; M1, presence of distant metastasis.
AJCC, American Joint Committee on Cancer; ENS@T, European Network for the Study of Adrenal Tumors; UICC, Union for International Cancer Control.
Prognosis
Although the overall prognosis of ACC is poor, survival can be heterogenous depending on the extent of disease. The majority of patients with ACC present with advanced disease (34% with stage III disease and 26% with stage IV disease) with the most common sites of metastatic spread being the liver, lungs, lymph nodes, and bone.5,30 Typically quoted 5-year survival rates (in European and North American cohorts) are 66–82% for stage I, 58–64% for stage II, 24–50% for stage III, and 0–17% for stage IV disease.3,71,72,79 A more recent Finnish series reported favorable 5-year survival rates of 100%, 93%, and 63% for stage I, II, and III disease, respectively. 32 This may reflect the increasing utilization of surgery and adjuvant therapy (discussed below) compared with historic studies. Despite this, 5-year survival remained dismal (11%) for patients with stage IV disease. 32
Targeted molecular classification as a future tool for individualized prognostication and management
As discussed above, in addition to providing insights into the pathogenesis of ACC, large-scale multi-omics studies from the ENS@T 7 and TCGA-ACC 8 groups have defined distinct subgroups of patients with clinically significant differences in survival based on genomic, epigenomic, and transcriptomic signatures. However, the requirement for prospective collection of fresh-frozen tissue and the costly and resource-intensive nature of pan-genomic bioinformatics analysis precludes its use in routine clinical practice. 80 As a method of overcoming these limitations, two recent studies have described how targeted molecular profiling (i.e. analysis of only a limited number of the most predictive biomarkers) could recapitulate the prognostic classification provided by the more comprehensive ENS@T and TCGA-ACC studies at a fraction of the required time and cost (Figure 3).81,82 Furthermore, in one of these studies, 81 targeted analysis was feasible using only formalin-fixed paraffin-embedded (FFPE) tissue, which is readily available in most clinical settings. Importantly, the combination of targeted molecular data with clinicopathological parameters in both studies was superior in predicting RFS compared to either component alone. In the future, increased utilization of targeted molecular analysis raises the possibility of individualized prognostication and precision medicine in patients with ACC, as has been implemented for other cancers. 83
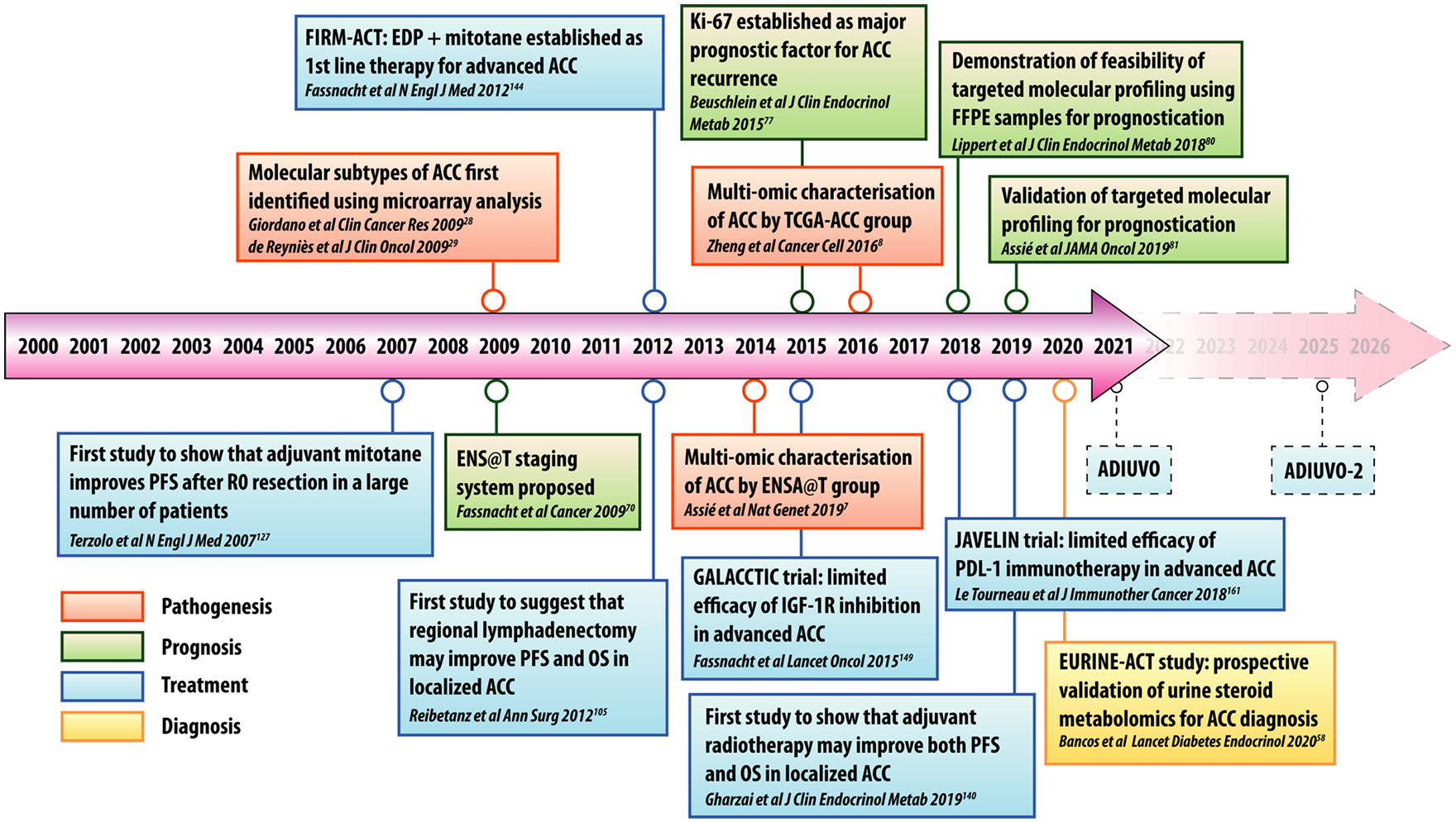
Timeline of important studies in the pathogenesis, diagnosis, treatment, and prognostication of ACC over the past 20 years.
Treatment
Complete surgical resection with negative margins (R0) is currently the only curative treatment for ACC and applies to patients with resectable stage I–III disease. Adjuvant therapies are used to decrease recurrence rates, which are reported to range from 40–70% even after R0 resection, depending on stage.4,84,85 For patients with unresectable stage III or metastatic (stage IV) disease, the goals of treatment are palliative rather than curative. An additional treatment challenge involves counteracting the deleterious effects of excess hormone secretion, which is associated with increased morbidity and impaired quality of life. For these reasons, and the fact that most patients present with advanced disease, it is strongly recommended that patients with ACC are managed by multidisciplinary teams at institutions with experience and expertise in treating this rare malignancy. 86 An overview of current emerging treatment options for patients with localized and advanced disease are provided below. The management of hormone excess in ACC is beyond the scope of this review, and has been extensively covered by Else et al. 30
Surgery
Because the only chance for cure in patients with ACC is afforded by complete resection, surgery may necessitate en bloc removal of not only the tumor, but also any adjacent involved organs, such as the ipsilateral kidney, pancreas, and/or diaphragm, as well as the periadrenal retroperitoneal fat, in order to achieve an R0 resection. 6 In the absence of direct invasion, however, there is no convincing evidence that routine en bloc resection of extra-adrenal organs is associated with superior oncological outcomes when negative margins can otherwise be achieved.87,88 Regardless of operative extent, it is crucial that surgery is performed meticulously to avoid intraoperative tumor rupture and spillage, which are associated with high recurrence rates and extremely poor survival. Extension of tumor thrombus into the inferior vena cava (T4 disease) is not a contraindication to surgery and may require cross-clamping of the retro-hepatic inferior vena cava and/or cardiopulmonary bypass. 89 However, this technique requires careful patient selection as long-term survival outcomes remain suboptimal in this subgroup. 90
The nuances and challenges of ACC surgery underscore the strong recommendations in current guidelines for these procedures to be performed at specialized centers by experienced adrenal surgeons.5,6 Indeed, a volume-outcome effect in adrenalectomy has been repeatedly described, with fewer complications reported when procedures are performed by expert surgeons at higher volume centers. 91 Whether this translates into improved oncological outcomes in ACC has not consistently been shown, however, with some studies suggesting longer time to recurrence 92 and better overall survival 93 when surgery is performed at high- versus low-volume centers, while others have failed to demonstrate any survival advantage. 94 Furthermore, the specific number of ACC surgeries that defines a ‘high-volume’ surgeon or center is unclear and varies widely in the literature from 4 to 20 cases per year.6,92,95
Choice of surgical approach
Despite the benefits afforded by minimally-invasive adrenalectomy in terms of reduced blood loss, improved cosmesis, and shorter recovery time, its role in ACC surgery remains controversial.96 –100 This is due to the concerns that achieving a curative margin-negative resection may be more challenging laparoscopically, particularly in cases where an infiltrative tumour obliterates dissection planes and precludes good exposure. 6 A thin tumour capsule (e.g. in cortisol-secreting ACCs) may also be more likely to rupture secondary to manipulation with laparoscopic instruments. In accordance with this, some reports have shown higher rates of ACC recurrence (including in port sites) and poorer disease-free survival with a laparoscopic approach.101,102 However, the literature is not unequivocal on this topic and, as additional experience has accumulated, comparable oncological outcomes with laparoscopic and open adrenalectomy for ACC have also been reported, particularly for smaller tumors without evidence of local invasion.103,104 The lack of consensus regarding the role of laparoscopy is reflected in differences between guidelines from the American Association of Endocrine Surgeons (AAES)/American Association of Clinical Endocrinologists (AACE) which recommend open resection for all cases of ACC (regardless of size) 105 versus newer ENS@T/ESE guidelines that suggest that laparoscopic resection is a reasonable option for localized tumors <6 cm in diameter. 6
The role of lymphadenectomy
Locoregional lymphadenectomy for therapeutic purposes is standard practice for many intra-abdominal malignancies; however, its role in ACC is poorly defined. The first study that attempted to address this topic retrospectively analyzed 283 patients from the German ACC Registry with stage I–III disease, 17% of whom underwent lymph node dissection (defined as the removal of ⩾5 lymph nodes) during curative-intent adrenalectomy. 106 On multivariable analysis, lymph node dissection was independently associated with a 35% lower risk of tumor recurrence and a 46% lower risk of disease-related death. 106 A subsequent retrospective US multi-center study evaluated 120 non-metastatic ACC patients, 27% of whom underwent lymphadenectomy (defined as a documented attempt to dissect regional lymph nodes in the operative note). 107 The authors found that lymphadenectomy was associated with a significantly higher 5-year OS rate of 76% compared to 59% in the group that had no lymph nodes excised. 107 However, multiple studies using the SEER registry and National Cancer Database (NCDB) have shown contradictory results, with either no effect, 108 worse survival,109 –112 or a marginal improvement in survival, 113 in patients who underwent lymphadenectomy. Reasons for these discrepancies include the fact that the inclusion criteria in these studies differ widely by stage, presence of metastases, multivisceral resection, and margin status. In addition, the lack of granularity intrinsic to datasets such as SEER and NCDB make it impossible to determine whether lymphadenectomy was performed intentionally, and which specific lymph node basins were removed. Collectively, these studies highlight the need for a prospective trial in which the a priori objective is to determine whether lymphadenectomy confers a survival benefit in ACC patients. A recent study that described radiological patterns of regional nodal recurrences in 56 ACC patients suggested that the definition of lymphadenectomy that is used in a future prospective trial should include removal of periadrenal, renal hilar, ipsilateral para-aortic, or paracaval nodes. 114
Surgery for locally recurrent or metastatic disease
Approximately 20–60% of reported recurrences in ACC patients are locoregional. 115 In these cases, reoperation may be indicated if an R0 resection is achievable. This is supported by data from a number of retrospective studies, which have shown improvements in symptoms and a median survival of >5 years in selected patients who undergo reoperation versus medical management alone.115 –119 Studies have also demonstrated the safety and efficacy of pulmonary 120 and hepatic metastatectomy, 121 with reported 5-year OS rates of ~25–50%. Overall, patients who derive the greatest benefit from reoperation appear to be those with a disease-free interval from initial resection to recurrence of >9–12 months.117,122,123 The decision to offer reoperation should weigh up the potential survival benefits against the risks of morbidity (12–55%), mortality 0–4%), and the high likelihood of further recurrence (~80%). 6
The role of metastasectomy in patients who have synchronous metastases at presentation is less clear, with studies showing that these patients have a poorer overall prognosis compared to those who develop metachronous metastases.124,125 Studies describing operative intervention in this group are lacking due to the paucity of cases and short duration of survival. A combined series of 27 patients from Mayo Clinic and the MD Anderson Cancer Center showed that those who underwent an R0 resection had significantly better median OS of 28.6 months versus 13.0 months when only an R2 resection was acheived. 126 A similar benefit was also reported in another US multi-center study of 26 patients with Stage IV disease (median survival of 19.0 months for R0 resection versus 5.5 months for R1/R2 resection). 125 Overall, these findings indicate that patients with metastatic ACC constitute a heterogenous group with distinct differences in tumor biology, prognosis, and degree of benefit derived from surgery.
Adjuvant therapy
Although surgical resection is technically feasible in most patients with stage I–III ACC, the majority of patients still succumb to disease, presumably due to occult micrometastases that are present at the time of initial presentation. As a result, adjuvant therapies may be used in an attempt to reduce rates of disease recurrence in patients who have a seemingly curative index operation.
Mitotane
Mitotane is a derivative of the insecticide dichlorodiphenyltrichloroethane (DDT) and exerts adrenolytic activity through mechanisms that are incompletely understood. One of these mechanisms includes endoplastic reticulum stress activation, which impairs steroidogenesis and induces apoptosis of ACC cells. 127 Impetus for the use of mitotane in the adjuvant setting arose from a 2007 retrospective study by Terzolo et al., 128 which analyzed 177 stage I–III ACC patients who were followed up for up to 10 years at referral centers in Italy and Germany (Figure 3). Adjuvant mitotane use was associated with significantly longer median recurrence-free survival of 42 months versus 10 and 25 months in the Italian and German control groups, respectively. Median OS was also better in the mitotane group at 110 months versus 52 and 67 months in the Italian and German cohorts, respsectively. 128 A subsequent description of this cohort, after almost 10 additional years of follow-up, showed a sustained survival benefit in the mitotane group. 129 A more recent study from this group of authors, in an independent sample of 152 patients, showed that adjuvant mitotane prolonged recurrence-free survival without improving overall survival, although better OS was seen in a subgroup of patients deemed to be at high risk of recurrence (elevated Ki-67 and stage III disease). 130
In contrast, studies from US centers have shown conflicting results regarding the benefits of adjuvant mitotane on survival outcomes.131 –133 The University of Michigan group observed that while adjuvant mitotane significantly improved RFS, no effect on OS was seen. 131 In a study from MD Anderson, patients who underwent index surgery at that institution had a recurrence rate of 50% after a median follow-up of 7.3 years, 132 which the authors noted was similar to the 49% 5-year RFS rate reported for patients who received adjuvant mitotane in the 2007 Italian/German study by Terzolo et al. 128 Because 90% of the MD Anderson cohort did not receive adjuvant mitotane, the authors attributed the comparable recurrence rates in their study to the quality and completeness of surgery performed at their institution, rather than to the use of adjuvant therapy. Similarly, in a 13-institution study from the US ACC group, no significant association between mitotane use and RFS or OS was seen on multivariable analysis. 133 It is important to note that all of the aforementioned studies are retrospective in nature and are fraught with selection bias and heterogeneity with regards to patient characteristics and treatment regiments. While recognizing that the evidence base is weak, current European guidelines recommended adjuvant mitotane for patients who are at high risk of recurrence (e.g. stage III disease, R1 resection, or Ki-67 > 10%).5,47 Whether or not adjuvant mitotane is beneficial in patients at low or moderate risk of recurrence (e.g. stage I–II disease, R0 resection, and Ki-67 < 10%) is unclear, and it is anticipated that the recently-completed Efficacy of Adjuvant Mitotane Treatment (ADIUVO) randomized controlled trial (RCT) will answer this question (Figure 3). Furthermore, the ongoing ADIUVO-2 RCT (due to complete in 2025) will determine whether the combination of cisplatin and etoposide with adjuvant mitotane is more effective than adjuvant mitotane alone in prolonging RFS and OS in patients with high-risk ACC (Figure 3).
Radiotherapy
Adjuvant radiotherapy is infrequently used in the management of ACC, with only 6 and 14% of patients reported as receiving this treatment modality in two separate NCDB studies covering the periods 1985–2005 134 and 2004–2013, 135 respectively. Reluctance to refer patients for radiotherapy likely reflects the widely held viewpoint that ACCs are radioresistant tumors, which is based on data from small pre-2000 series which failed to show any improvement in recurrence or overall survival with adjuvant radiation.136 –138 However, the results of recent studies that utilize modern radiotherapy techniques have challenged this notion.139 –141 For example, in a case-control study from the German ACC group, in which 14 patients with non-metastatic ACC who received adjuvant radiotherapy were matched to 14 patients that did not, local recurrence was observed in 14% of patients in the radiation group versus 79% of patients in the control group. 139 However, no significant differences were seen in terms of disease-free survival and OS. Findings consistent with these were reported by the University of Michigan group, in a case-control study of 20 patients who received adjuvant radiotherapy versus 20 matched controls who did not. 140 In this study, 5% of the patients who received radiation experienced a recurrence versus 60% of patients in the control group. Again, no significant differences in RFS or OS were found. The authors speculated that the lack of a detectable survival benefit was due to the study being underpowered and have recently published a larger series of 78 patients, half of whom received adjuvant radiation. 141 Interestingly, in addition to improved local recurrence rates, this study reported significantly better RFS and OS in the radiation group. It should be noted that there are no prospective data evaluating the benefit of adjuvant radiation, and the findings of retrospective studies should be carefully interpreted in light of the potential for referral and selection bias. Despite this, emerging evidence suggests that adjuvant radiotherapy in the contemporary era is probably more effective than previously thought.
Systemic therapy
The dearth of effective systemic therapies poses a significant challenge in the treatment of patients with metastatic ACC. For years, mitotane has remained the only drug approved by the United States Food and Drug Administration (FDA) and the European Medicines Agency (EMA) for the treatment of unresectable ACC. 142 For selected patients with low tumour burden and/or slower growth kinetics, mitotane monotherapy has been used, albeit with disappointing objective RR of 20–25%.143,144 Otherwise, standard of care palliative treatment is with etoposide, doxorubicin, and cisplatin, in combination with mitotane (EDP-M). Evidence in support of this regimen came from the FIRM-ACT (First International Randomized Trial in Locally Advanced and Metastatic ACC Treatment) study (Figure 3). 145 In this phase III RCT, the authors compared EDP-M (n = 151 patients) with another commonly used regimen at the time, streptozosin plus mitotane (SZ-M; n = 153), demonstrating superior efficacy in the EDP-M arm (23% RR) compared with the SZ-M arm (9% RR). 145 Progression-free survival (PFS) in the EDP-M arm was also significantly longer at 5 months versus 2 months in the SZ-M arm, and there was a suggestion of better OS in the former group (14.8 months for EDP-M versus 12 months for SZ-M), although this did not reach statistical significance, possibly due to the cross-over design of the trial. While this study was important in establishing the superiority of EDP-M for ACC patients with metastatic disease, it also highlighted that, despite gold-standard treatment, prognosis in this group of patients remains dismal.
For patients who experience disease progression despite EDP-M, there is limited evidence regarding the efficacy of non-first line chemotherapies. The two most commonly studied second-line agents include SZ-M and gemcitabine plus capecitabine, with or without mitotane.146,147 However, RRs with these regimens have been disappointing (<10%). A recent study investigated temozolomide as a second- or third-line treatment for metastatic ACC showing a RR of 20%, although this was short-lived and did not influence median OS, which remained poor at 7.2 months. 148
Newer treatments: targeted therapies, immunotherapy, and radiopharmaceuticals
Given the limited effectiveness of current treatments, the search for novel therapeutic strategies for advanced ACC is a current research priority. Findings from the comprehensive ENS@T 7 and TCGA-ACC 8 molecular profiling studies have provided an exhaustive road map of potential druggable targets. Unfortunately, however, this knowledge has not yet resulted in a treatment that demonstrates efficacy above what is currently used in the clinic. For example, the observation that IGF2 is overexpressed in the majority of ACCs and that inhibition of IGF2/IGF1R was effective in reducing tumor growth in pre-clinical models 149 led to the evaluation of lisitinib (a small molecule inhibitor of both IGF1R and the insulin receptor) in a phase III RCT, the GALACCTIC (A Study of OSI-906 in Patients with Locally Advanced or Metastatic Adrenocortical Carcinoma) trial (Figure 3). 150 Disappointingly, no differences in OS or RFS were seen compared to placebo. In addition, only 15.6% of patients in the lisitinib arm achieving disease control, with a median PFS survival of <2 months. Other targeted therapies that have been, or are currently undergoing, evaluation in phase I/II trials include epidermal growth factor receptor (EGFR) inhibitors, 151 vascular endothelial growth factor receptor (VEGFR) inhibitors, 152 fibroblast growth factor receptor (FGFR) inhibitors, 153 multityrosine kinase inhibitors (TKIs),154,155 and sterol-O-acetyltransferase (SOAT1) inhibitors. 156 Unfortunately, these compounds have shown limited efficacy, with temporary disease stabilization observed in best-case scenarios. Small molecule inhibitors that have been tested pre-clinically but have not yet been evaluated in early-phase trials include inhibitors of the Wnt/βcatenin signalling, 157 cyclin-dependent kinases, 158 Notch signalling, 81 and mitogen-activated protein kinase (MAPK)/ERK signalling. 159 Preclinical data supporting the rationale for their use in ACC have been reviewed recently by Altieri et al. 160
Immunotherapy has emerged as a standard pillar of treatment across a broad spectrum of solid tumors, 161 which was spurred enthusiasm for investigating this class of treatment in ACC. To date, four phase I/II clinical trials of immune checkpoint inhibitors in ACC have been published.162 –165 The largest of these, the Avelumib in Metastatic or Locally Advanced Solid Tumors (JAVELIN) trial, was a phase Ib study that evaluated the efficacy of avelumab [a programmed death-ligand 1 (PD-L1) antagonist] in 50 patients with metastatic ACC who had failed to respond to platinum-based chemotherapy (Figure 3). 162 Disappointingly, the overall RR was only 6% with a median PFS survival of 2.6 months. Potential explanations for the modest results seen in immunotherapy trials in ACC include an immunosuppressive action of locally secreted glucocorticoids, and deregulated Wnt/β-catenin signaling, leading to impaired immune cell infiltration. 166
As described above, a subset (~30%) of patients with metastatic ACC demonstrate significant uptake of MTO in both the primary tumor and distant metastases. 53 By replacing [ 123 I] with [ 131 I], MTO has been adapted for targeted radionuclide therapy. 167 In a preliminary study of 11 patients with advanced ACC who demonstrated [ 123 I] MTO avidity on diagnostic scans, a partial response to 1–3 cycles of [ 131 I] IMTO was achieved in 1 patient, with stable disease achieved in 5 patients. 167 Based on the observation that some ACCs may express somatostatin receptor (SSTRs), 168 peptide receptor radionuclide therapy (PRRT) using the radiolabeled somatostatin analogues [ 90 Lu]- and [177Y]-DOTATOC was recently reported in a preliminary study of 19 patients by Grisanti et al. 169 In this study, 2 patients were selected for PPRT based on the finding of strong uptake of [ 68 Ga]-DOTATOC on PET/CT, indicating high somatostatin receptor expression. Both patients experienced disease stabilization that lasted 4 months and 12 months, respectively. 169 As a result, radiopharmaceutical therapy may represent a potential salvage treatment option for a small subset of cases of metastatic ACC, although this requires validation in a larger number of patients.
Conclusions and future perspectives
ACC is a rare and devastating malignancy that entails a poor prognosis in the majority of cases. The past 20 years have seen the publication of a number of important studies that have provided insights into the pathogenesis of ACC and its diagnosis and treatment (Figure 3). In patients with adrenal incidentalomas, the diagnosis of ACC may be challenging due to the limited diagnostic accuracy of cross-sectional imaging. Implementation of urine steroid metabolomics into routine workup could improve detection rates and spare patients with benign masses from undergoing unnecessary surgery, although the cost-effectiveness of this technology remains to be demonstrated. Large-scale pan-genomic analyses have greatly improved our understanding of the pathogenesis of ACC and have paved the way for targeted molecular profiling in the clinic with the possibility of individualized prognostication to guide therapy in the future. Surgery remains the only curative treatment for ACC, although questions remain about the optimal surgical approach, the benefits and definition of regional lymphadenectomy, and the effects of surgeon volume on oncological outcomes. Despite seemingly curative resection, most patients experience disease recurrence and a better understanding of the factors that influence prognosis and response to adjuvant mitotane treatment are required. Results from ADIUVO and ADIUVO-2 will provide valuable insights into this topic. The role of postoperative radiation needs to be better defined in prospective studies, as emerging evidence suggests a survival benefit in selected patients. There is a clinically unmet need for improved treatments for advanced ACC, as current standard of care is associated with suboptimal survival. Trials of molecularly targeted therapy and immunotherapy have been disappointing to date, suggesting that combination therapies that are guided by molecular biomarkers may be more successful than a blanket ‘one-size-fits-all’ approach. Given the rarity of ACC, the success of future research efforts is contingent upon continued international collaborations between teams that recognize the urgent need to widen treatment options and improve outcomes for patients with this orphan disease.