
Abstract
Intravenous infusion of alpha-1 antitrypsin (AAT) was approved by the United States Food and Drug Administration (FDA) to treat emphysema associated with AAT deficiency (AATD) in 1987 and there are now several FDA-approved therapy products on the market, all of which are derived from pooled human plasma. Intravenous AAT therapy has proven clinical efficacy in slowing the decline of lung function associated with AATD progression; however, it is only recommended for individuals with the most severe forms of AATD as there is a lack of evidence that this treatment is effective in treating wild-type heterozygotes (e.g., PI*MS and PI*MZ genotypes), for which the prevalence may be much higher than previously thought. There are large numbers of individuals that are currently left untreated despite displaying symptoms of AATD. Furthermore, not all countries offer AAT augmentation therapy due to its expense and inconvenience for patients. More cost-effective treatments are now being sought that show efficacy for less severe forms of AATD and many new therapeutic technologies are being investigated, such as gene repair and other interference strategies, as well as the use of chemical chaperones. New sources of AAT are also being investigated to ensure there are enough supplies to meet future demand, and new methods of assessing response to treatment are being evaluated. There is currently extensive research into AATD and its treatment, and this chapter aims to highlight important emerging treatment strategies that aim to improve the lives of patients with AATD.
Keywords
Introduction
At present there is only one disease-modifying pharmacological intervention available specifically for the treatment of alpha-1 antitrypsin deficiency (AATD): intravenous (IV) infusion of alpha-1 antitrypsin (AAT). There are several products currently available in the United States (US) to treat emphysema in patients with AATD that have been approved by the US Food and Drug Administration, and there are three approved products currently available in Europe (Table 1).1,2 Each product is prepared from pooled human plasma. However, as discussed in previous chapters in this series of reviews, this IV AAT therapy has limitations for its use, such as cost implications and patient burden.3,4 Therefore, patients with AATD have substantial unmet needs and several new on-going developments are focusing on reducing patient inconvenience, while providing efficacy in decreasing morbidity and mortality. 5 Following issues raised by patients on the need for a global registry, further information on comorbidities, natural history of the disease, risk factors for progression and the poor prognosis of lung disease, the European Alpha-1 Research Collaboration (EARCO) was created. 6 The EARCO is a Clinical Research Collaboration of the European Respiratory Society, which will collaborate with the European Reference Network (ERN) for respiratory diseases (ERN-LUNG), the ERN for hepatological diseases (ERN RARE-LIVER), and the registry of the Alpha-1 Liver Group. 6 The aim of the collaboration is to bring together researchers, healthcare providers, patients, and industries to advance our understanding and improve the quality of life for patients with AATD.6,7
Approved AAT therapy preparations.

Aralast-NP is a newer preparation of a previously approved product, Aralast™. 17
Prolastin®-C is a newer preparation of a previously approved product, Prolastin®. 18
Zemaira® is marketed as Respreeza® in Europe.
Prolastin® is licensed in the member states of European Economic Area in Austria, Ireland, Italy, Germany, Greece, Poland, Portugal, and The Netherlands. Prolastin® is also marketed as Prolastina® in Denmark, Finland, Norway, Spain, and Sweden, and marketed as Pulmolast® in Belgium.
AAT, alpha-1 antitrypsin; EU, European Union; US, United States.
The current chapter in this series of reviews on AATD aims to raise awareness of these current unmet needs and new therapy developments currently in the pipeline, including use of biomarkers to evaluate disease progression and treatment response to disease-modifying intervention. However, as AATD is a rare disease, it can be challenging to obtain conclusive data from small patient cohorts. 8 Nevertheless, rare disease drug development is an area that is rapidly expanding and there is a trend between the increasing number of clinical trials and the increasing number of rare diseases being reported, 9 with many technologies for treatment available. 10 Several of these technologies are discussed herein with relation to their use within the field of AATD.
Current treatment and unmet needs
The only licensed treatment that has a disease-modifying effect on AATD progression is weekly IV infusion of plasma-derived AAT, which aims to delay the progression of emphysema,19,20 reduce exacerbation frequency, 21 and improve patient quality of life.5,22 Further information on the current treatment recommendations for AATD can be found within the chapter of this review series authored by Barjaktarevic and Campos. 4 However, not all countries have accepted AAT therapy. Current IV AAT therapy incurs significant annual healthcare costs, which exceed prior estimates reported in a 2003 study on cost effectiveness for patients with severe AATD. 23 In the US, AAT therapy costs an estimated US$82,000 per patient per year, excluding other out-of-pocket costs to the patient, such as visits to the physician and/or emergency department, inpatient hospital stays and other prescription drug costs. 24 The mean annual out-of-pocket cost for all patients with AATD is approximately US$1875, with annual costs of US$4601 and US$1689 for IV AAT therapy users and non-users, respectively. 24 As AAT therapy is lifelong, treatment is costly and is commonly used only to treat the most severe forms of AATD (patients with PI*ZZ and PI*Null genotypes). This treatment has also been shown to change imaging characteristics of AATD based on evidence from the RAPID program.19,20 However, patients with less severe forms of AATD (e.g., PI*MZ and PI*SZ genotypes) can still experience disease symptoms but are often left untreated. Furthermore, the prevalence of these intermediate forms of AATD is thought to be greater than initial estimates, 25 suggesting that there are even more individuals who may possibly benefit from therapy that go untreated. There is a rationale to treat these patients once a more widely available therapy has shown efficacy.
At present, IV AAT therapy regimens can be inconvenient for patients due to the need for lifetime weekly infusions. 5 Moreover, this therapy addresses only AATD-associated lung disease and does not treat extra-pulmonary manifestations such as liver disease, which is discussed in detail within the chapter by Patel and Teckman. 26 Therefore, new cost-effective treatments are being sought that would not only be more convenient for patients, but also address the underlying genetic component of AATD, rather than simply slowing disease progression and reducing symptom severity. More cost-effective therapy may encourage further research, adoption of therapy by more countries, and allow treatment of patients with less severe forms of AATD who are currently untreated.
Research and future treatments
Sourcing AAT
Supply of pooled human plasma-derived AAT can be variable in purity and activity. 27 Therefore, new methods of obtaining AAT are required to improve consistency and to ensure supplies are able to meet future therapy demands. The human AAT gene, SERPINA1, has been expressed successfully in bacteria, 28 yeast, 29 and mammalian cell lines,30,31 as well as in animal models,32,33 but to date no therapeutic product has been licensed from these alternative sources. New products obtained via alternative methods would need to meet several requirements for licensing. New products should be clinically efficacious, safe, demonstrate good plasma half-life, and be cost-effective relative to current sources of AAT. Due to the potential risk of contamination with new and unknown pathogens from current plasma-derived AAT, recombinant versions of AAT (rAAT) are also an attractive alternative that is being widely investigated. Modified rAAT is currently being explored in fusion protein-based therapy to treat patients with AATD with the aim of offering superior clinical activity to plasma-derived AAT by maintaining plasma concentration with a less frequent, monthly, dosing regimen. 34
Escherichia coli is a readily available and common recombinant protein expression platform for a wide variety of proteins due to its fast growth kinetics, inexpensive growth media, and simplicity of transformation with exogenous DNA, despite its inability to glycosylate recombinant proteins. 35 Lack of protein glycosylation in the E. coli expression system can affect rAAT activity, circulating half-life after infusion, and cause rAAT aggregation, 36 as plasma-derived human AAT contains three glycosylation sites important for protein stability and protection from proteolysis and/or degradation. 37 The lack of rAAT glycosylation could potentially be overcome by utilizing genetically modified E. coli that have proven to be 100% efficient in glycosylating rAAT. 38
Plant cell suspension systems are also emerging as a promising alternative for bioproduction of pharmaceutical agents, including AAT, and are able to express high yields of glycosylated rAAT in relatively short periods of time.39,40 However, there is currently a lack of regulatory approval for plant expression systems. 41 Yeast systems are another attractive option for rAAT production due to their cost and time efficiency. 42 Yeast systems may also be more advantageous over bacteria as they do not produce endotoxins and are able to provide some post-translational modifications, including glycosylation. However, a mammalian system would be ideal in expressing a rAAT more akin to human AAT. Transduction of mammalian cell lines with a lentiviral gene delivery system can be successful in producing large amounts of recombinant human AAT 30 but, unfortunately, mammalian expression systems are complex, costly, time-consuming and associated with limitations in protein yield. 43
Inhalation AAT therapy
Early studies have reported that, in addition to the expense and inconvenience of IV AAT therapy, only 2–3% of IV AAT reaches the lungs. 44 Therefore, inhalation therapy has been studied in order to deliver AAT directly to the lungs; a method that has attracted a high level of interest from patients with AATD. 45 A study from as far back as the late 1980s demonstrated that inhaled AAT can increase AAT levels in the lung epithelial lining fluid in patients with severe AATD (PI*ZZ) from 0.28 µM to 5.86 µM. 46 Since then, little has been reported on aerosolized AAT therapy in patients with AATD, and IV AAT therapy has been the standard method of administration. However, as we are now looking to further improve the lives of patients with AATD, 5 inhalation therapy is gaining more momentum.
In 2001, it was shown that more AAT reaches the lungs via inhalation than through the IV route (14.6% versus 2%, respectively), 47 further confirming that inhalation therapy may be a more convenient route of AAT administration. It has also been suggested that inhalation therapy may lead to improved mobility and improved quality of life for patients who are currently restricted by the need for frequent IV AAT infusions. 47 The improved delivery of AAT via the lungs may also reduce healthcare costs. 47 However, there are concerns about clinical efficacy of inhalation as a delivery method because in 2019 a clinical trial suggested there was no associated reduction in exacerbation frequency. 48 The randomized placebo-controlled clinical trial was conducted in 168 patients with AATD (PI*ZZ) and severe chronic obstructive pulmonary disease (COPD), who experienced frequent exacerbations. Results demonstrated that AAT inhalation therapy had no effect on the time to first exacerbation during 50 weeks of treatment. 48 The time to the first moderate/severe exacerbation was a median of 112 days for patients receiving inhaled AAT and 140 days for placebo treatment. 48 Furthermore, patients receiving AAT reported more treatment-related adverse events (such as dyspnea, cough, respiratory tract infection, and nausea) compared with placebo (57.5% versus 46.9%, respectively). 48 Following improvements in the handling of the AAT and nebulizer, the safety profile of the AAT treatment group became similar to that of the placebo group, resembling the results reported in the IV AAT study (RAPID 19 ). 48 Nevertheless, a prospective phase III trial is currently underway to determine the efficacy and safety of AAT inhalation therapy in patients with AATD. 49 The trial is expected to end in 2023 and is being conducted in patients with AATD who have moderate airflow limitation [forced expiratory volume in 1 s (FEV1) ⩽50–80% of predicted] and an FEV1/slow vital capacity ⩽70%. 49 At least 220 patients are expected to be enrolled in the study. After 4 weeks of practicing with the nebulizer, patients will be randomized to either 80 mg/day AAT inhalation or placebo for 104 weeks. 49 During the study, inhaled AAT will be evaluated by lung function assessment via blood tests and computed tomography (CT) densitometry. 49 Information on the utility of CT densitometry in AATD studies can be found in this series’ chapter on imaging in AATD by Huang et al. 50
Epigenetics and genetic modifiers
The variable nature of AATD and its strong influence by environmental exposures and lifestyle factors implicate a major role for epigenetic regulation in the pathophysiology of AATD. Epigenetic traits are stably heritable phenotypes resulting from changes in chromosomes without alterations in DNA sequence. 51 DNA methylation is an epigenetic trait usually associated with silencing gene expression, 52 and there is evidence of altered methylation patterns in patients with AATD, particularly in smokers. In a study of 316 patients with AATD (PI*ZZ), methylation of 16 methylation sites demonstrated significant association with ever-smoking and a younger age at smoking initiation, highlighting the harmful effect of smoking for patients with AATD. 53 Examples of epigenetic changes that are affected by smoking are shown in Figure 1.

Epigenetics in smoking-associated lung disease. Epigenetic modifications for cigarette smoking-related pulmonary biological activity mostly occur through DNA methylation, histone modification, and microRNAs.
Cigarette smoke is particularly harmful in patients with severe AATD (e.g., PI*ZZ). Cigarette smoke induces oxidation of AAT and accelerates polymerization of mutant AAT, inactivating the anti-elastase function whilst simultaneously creating a proinflammatory environment that reduces pulmonary defenses and increases neutrophil influx into the lungs. 55 Blocking cigarette smoke-induced oxidation with N-acetyl cysteine, a potent antioxidant, has been shown to prevent oxidation-induced polymerization of mutant AAT, potentially suggesting a novel anti-oxidative therapeutic strategy for treating AATD. 55
Modifier genes have also been reported for AATD. Two examples of genes that have been shown to be associated with lung function phenotypes in patients with AATD are cholinergic receptor nicotinic alpha 3 subunit (CHRNA3), which encodes a ligand-gated ion channel involved in neurotransmission, and iron responsive element binding protein 2 (IREB2), an RNA-binding protein involved in iron homeostasis. In the AAT Genetic Modifiers Study, single nucleotide polymorphisms (SNPs) in both CHRNA3 and IREB2 were shown to be associated with different lung function phenotypes in individuals with AATD, all of whom had the PI*ZZ AATD genotype. 56 The AAT Genetic Modifiers Study also suggested a potential sex-specific effect of these SNPs as they were significantly associated with lung function phenotypes in the male subgroup (p=0.02 and p=0.03 for CHRNA3 and IREB2, respectively), but not the female subgroup. 56
The vitamin D-binding protein (DBP) may also play a role in AATD. The DPG gene, group-specific complement (GC), is highly polymorphic, with three common variants and over 120 rare variants. 57 One of these DBP variants, the GC2 variant, which is less able to activate macrophages, has been shown to be associated with a decreased risk of COPD (p=0.05). 58 However, the same variant has been associated with an increased risk of bronchiectasis in patients with AATD (p=0.04). 58 Further studies will be required to ascertain whether vitamin D supplementation will be beneficial for patients with AATD.
Whole exome sequencing (WES), a technique for sequencing all protein-coding regions of genes within the genome, has now enabled further identification of genes that may be involved in the development or suppression of AATD. In a study of four families with severe AATD (PI*ZZ or PI*Z/Q0Brescia), WES identified 14 genes in a recessive model (PI*Z homozygosity) and 21 genes in a dominant model (PI*Z heterozygosity) of AATD that influence the risk of AATD development or avoidance. 59 Among these genes, of particular interest was the rs3747517 variant of interferon-induced with helicase C domain 1 (IFIH1), 59 a gene strongly associated with susceptibility to autoimmune diseases, such as type I diabetes and multiple sclerosis.60,61 However, a genome-wide association study (GWAS) of lung function has yet to be performed in patients with AATD. Such a study could potentially determine whether genetic factors of lung function in the general population overlap with those in AATD. 62 As the heterogeneity of lung function in AATD is partially attributable to genetics (which is further discussed in an earlier chapter of this review series), 63 further discovery of genetic modifiers of lung disease via GWAS could provide a greater understanding of AATD’s variable clinical presentation. 62
Emerging treatments
Gene repair strategies
Gene repair technology, such as CRISPR-Cas9 [clustered regularly interspersed short palindromic repeats (CRISPR) associated nuclease 9] is revolutionizing many areas of medical research, such as human immunodeficiency virus and hepatitis B virus infection, as well as monogenic diseases such as Duchenne muscular dystrophy. 64 CRISPR, therefore, represents an exciting approach that could be key to solving both lung and liver consequences of AATD (Figure 2). 65 In AATD mouse models, PI*ZZ mice show evidence of liver disease reversal upon successful gene editing via CRISPR, in addition to reduced mutant Z protein aggregation and restoration of modest levels of normal AAT expression.32,33
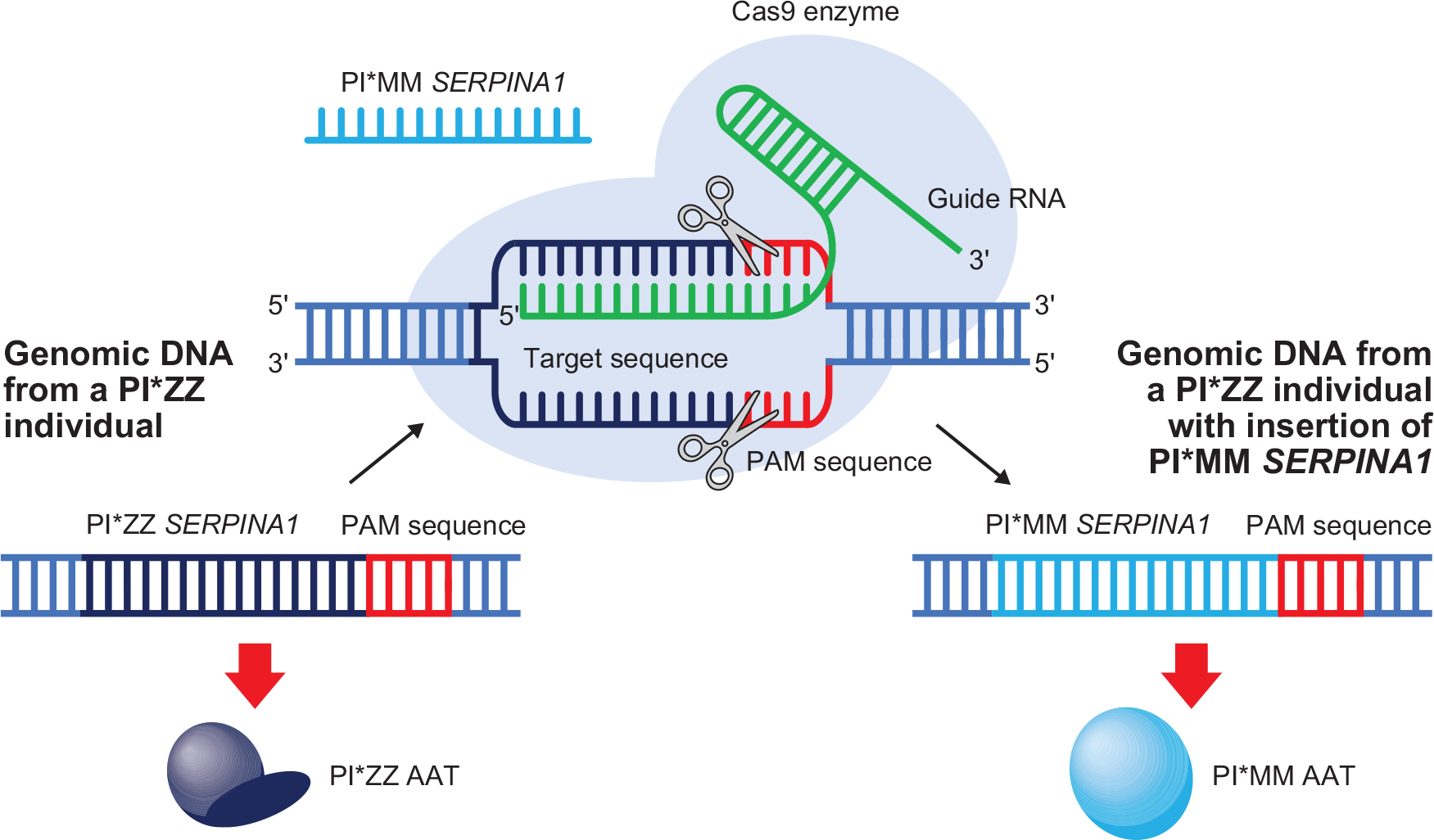
CRISPR-Cas9 to insert normal SERPINA1 into the genome of patients with the PI*ZZ gene mutation. The CRISPR-Cas9 method could be used to insert the gene for normal human AAT (SERPINA1) into the genomic DNA of patients with the PI*ZZ genotype. A guide RNA would target the insertion site, allowing the Cas9 endonuclease enzyme to cut host genomic DNA at specific PAM sequences. The repair sequence, in this case the normal SERPINA1, can then be inserted into genomic DNA of patients with PI*ZZ, allowing translation of normal PI*MM AAT.32,65
Besides CRISPR-Cas9, another method of gene therapy is the use of recombinant adeno-associated viral (rAAV) vectors to deliver the normal AAT gene (SERPINA1) to patients with PI*ZZ AATD. In a phase I clinical trial, rAAV vectors expressing normal AAT protein were injected intra-muscularly into nine patients with the PI*ZZ genotype. 66 The approach was associated with a positive safety profile and prolonged duration of normal AAT protein expression; however, serum AAT levels were more than 200-fold below the putative therapeutic target of 11 µM. 66 A phase II trial using a rAAV designed to generate a substantial increase in normal AAT expression still produced normal AAT levels below the therapeutic target. 67 Together, these studies show that rAAV-AAT vectors require further improvements to achieve target AAT concentrations required for therapeutic success.
An alternative to vector-based delivery of SERPINA1 is to use cell transplantation. In a PI*ZZ mouse model, engrafted wild-type hepatocytes spontaneously proliferated preferentially over host hepatocytes, replacing them by 20–98%, and this repopulation was accelerated by injection of an adenovector-expressing hepatocyte growth factor. 68 This method of hepatocyte transplantation could potentially be an effective therapy for patients with and without severe liver disease. 68
Human induced pluripotent stem cells (hiPSCs) derived from dermal fibroblasts from individuals with AATD are also being studied to advance the development of therapy for AATD. These cells have been used to generate patient-specific stem cell lines that have been differentiated into hepatocytes, enabling recapitulation of hepatic AAT protein folding to aid modeling of AATD’s pathological dysregulation of protein misfolding and therapeutic responses/toxicities to new pharmacological agents.69,70 hiPSCs have also been used in a proof-of-concept study on genetic correction, demonstrating that zinc finger nucleases in combination with the piggyBac gene editing technique can restore the normal structure, function, and secretion of AAT in subsequently derived hepatocytes, both in vitro and in vivo. 71
Chaperones and synthetics
As an alternative to gene therapy, use of chemical chaperones to improve proper folding of mutant AAT protein is being investigated. 4-phenyl butyrate (4PBA) was the most effective chemical chaperone in one study designed to increase the secretion of functionally active PI*ZZ AAT in a human cell culture model. 72 The chemical chaperone was also well tolerated in PI*ZZ mice, consistently increasing serum AAT levels up to 50% of those in normal humans and mice. 72 Furthermore, 4PBA is well tolerated in humans and therefore satisfies many of the criteria required for a potential chemoprophylactic tool for lung and liver injury in AATD. 72 On this basis, a pilot in-human trial was conducted, but no effect on AAT secretion was detected, likely due to an inability to safely achieve the therapeutic levels of 4PBA described in the PI*ZZ mouse model. 73 A major limitation of the chemical chaperone approach is the sheer mass of chemical chaperone required to be delivered to the hepatocyte endoplasmic reticulum. 74
In November 2019 a phase II trial was initiated to determine the efficacy and safety of facilitating proper folding of the PI*ZZ variant of AAT via oral delivery of an experimental protein chaperone (VX-814). 75 The oral delivery route has the benefit of being a less invasive drug delivery method than IV administration and is also associated with good patient compliance. 76 However, the VX-814 trial was terminated in October 2020 at the sponsor’s discretion as it was not feasible to safely reach targeted VX-814 exposure levels to provide a meaningful increase in levels of AAT. 77
Synthetic peptides are also being investigated for the treatment of AATD. A 6-mer peptide corresponding to the P7-2 sequence of the AAT reactive loop has been demonstrated to completely inhibit polymerization of the Z forms of AAT, without any inhibitory effect on the normal form of AAT. 78 This molecule therefore has potential to prevent the intracellular accumulation of Z AAT within hepatocytes and therefore treat AATD-associated liver disease. 78 However, this approach may result in the release of Z AAT proteins into the circulation and so would not address the need to increase levels of functional serum AAT; AAT replacement therapy would still be required.
Monoclonal antibodies have been developed to detect mutant conformations of AAT to help quantify levels of mutant AAT in biological samples and aid the development of small molecules to block polymerization.79,80 Intracellularly expressed antibody fragments (intrabodies) are emerging as one approach to modulate the function of targets in different intracellular compartments. 81 One such intrabody is scFv4B12, a single-chain variable fragment of the 4B12 monoclonal antibody that has been shown to prevent intracellular polymerization of PI*ZZ AAT and increase its hepatic secretion, while retaining neutrophil elastase inhibitory function. 82 Another intrabody has been developed that can not only inhibit the intracellular polymerization of Z AAT but also, when bound to Z AAT, allows the protein to retain almost two-thirds of its inhibitory activity against neutrophil elastase (NE). 82 The results with scFv4B12KDEL have been demonstrated only in vitro so-far but provide another step forward in the use of intrabodies for AATD treatment.
Finding a molecule that can bind to Z AAT, prevent polymerization, and restore NE inhibitory function both in vitro and in vivo is challenging, but the use of structure-based drug design is being employed to identify molecules that can bind to Z AAT in such a way to achieve this desired function. In silico ligand screening of 1.2 million small molecules has identified six compounds that reduced Z AAT polymer formation in vitro and 10 compounds that completely blocked PI*ZZ polymerization. 83 The lead compound identified in this study, CG, was also found to be highly selective and non-promiscuous, and therefore less likely to have unwanted off-target effects. 83
Interference strategies
Further strategies for future AATD therapy include several methods that interfere with AATD pathogenesis. RNA interference (RNAi) strategies include small interfering RNA molecules that can disrupt mutant AAT polymerization/degradation and manipulate autophagy for the treatment of liver disease in patients with AATD. Several applications of RNAi technology are currently underway with the aim of preventing synthesis of the Z form of AAT to prevent toxic hepatic accumulation and subsequent liver injury. Phase I/II and phase II/III clinical trials utilizing RNA silencing to prevent Z AAT protein synthesis as a therapy for AATD are now underway in Europe, 84 and the US. 85 These trials are expected to end in 2022 and 2023, respectively.
As discussed within the first chapter of this review series by Tejwani and Stoller, 3 the main damage inflicted on the AATD patient’s lung is through a proinflammatory imbalance of proteases, NE in particular. NE is one of the major proteases involved in a range of conditions, such as community-acquired and ventilator-associated pneumonia, acute lung injury and acute respiratory distress syndrome, exacerbated COPD, cystic fibrosis, and bronchiectasis. 86 In AATD, the involvement of NE and other proteases is rather complex, and the inability of IV AAT therapy to completely block emphysema progression in AATD suggests that alternative processes play a role in disease progression. 87 Nevertheless, NE inhibitors are being investigated for treatment of AATD. As a proof-of-concept study to investigate the mechanistic effect and the safety of an orally administered NE inhibitor, PI*ZZ/PI*SZ/PI*Null patients in the US are being treated over a period of 12 weeks, with an additional aim of investigating whether the treatment can reduce lung damage and slow disease progression. 88 The study is expected to be completed in August 2021. However, the role that other enzymes play in AATD-specific inflammatory processes warrants further investigation.
Biomarkers
Along with research into new treatments for AATD, there is also extensive research into new ways of testing AATD treatment efficacy, which is currently measured in the clinic using spirometry and CT-based lung density assessment. 89 In addition, biomarkers can also be reliable measures of treatment efficacy. Serum gamma glutamyl transferase (GGT) is frequently used as a biomarker of liver disease but may be particularly useful as a biomarker in AATD as it has been shown to be widely elevated in patients with the PI*ZZ genotype. 90 The relationship between serum GGT, lung disease, liver disease, and mortality in patients with AATD is independently associated with airflow obstruction and mortality. 90 Although not specific to AATD, in patients with COPD, several markers such as chemokine (C-C motif) ligand 18 (CCL-18), C-reactive protein, fibrinogen, interleukin (IL)-6, IL-8, surfactant protein D, and white blood cell counts are all elevated in non-survivors compared with survivors, and are specifically associated with patient mortality over 3 years. 91 In addition, elevated levels of plasma fibrinogen, another glycoprotein synthesized in hepatocytes, is associated with a risk of COPD and other inflammatory diseases.92,93 In AATD specifically, a fibrinogen degradation product (Aα-Val360) has been found to be increased in patients with AATD, and positively correlated with spirometric severity of AATD lung disease, as well as sputum elastase activity in acute exacerbations. 94 Furthermore, Aα-Val360 levels are decreased in patients receiving IV AAT therapy and it is, therefore, a potential biomarker of treatment response. 94
Levels of desmosine and isodesmosine (DES/IDES; markers of elastin degradation) are also elevated in AATD and non-AATD COPD and, like Aα-Val360, can be utilized as biomarkers of AAT therapy response. DES/IDES plasma levels have been shown to be significantly reduced in patients receiving AAT therapy (p<0.001) 95 and in bronchoalveolar lavage fluid from patients receiving double dose AAT therapy (p=0.050). 96 Levels of proteinase 3 (PR3) could be another potential biomarker for AATD. PR3, like AAT, is a serine proteinase and is released by neutrophil azurophilic granules along with NE following neutrophil activation. 97 Cleavage of fibrinogen with PR3 produces several fragments, one of which, Aα-Val541, can be detected in plasma from patients with AATD. 98 An enzyme-linked immunosorbent assay (ELISA) for Aα-Val541 performed in sera from patients with AATD (PI*ZZ and PI*SZ) has shown that levels of Aα-Val541 are much higher (~17 times higher) than those of Aα-Val360,98 and therefore it may be a more sensitive biomarker than Aα-Val360. The Aα-Val541 ELISA was also sensitive to augmentation therapy and therefore also represents a potential biomarker for AAT dose-ranging studies. 98
The tumor necrosis factor-alpha (TNF-α) gene represents a potential genetic biomarker for AATD. TNF-α is a pro-inflammatory cytokine that has been implicated in the pathophysiology of several pulmonary diseases, such as asthma, chronic bronchitis, and COPD. 99 Levels of inflammation have been demonstrated to be higher in patients with AATD compared with non-AATD-related COPD, 100 and AAT has also been shown to down-regulate TNF-α gene expression. 101 Wood and colleagues hypothesized that TNF-α may play a role in AATD; however, they found no association between TNF-α levels and disease severity as measured by lung function assessment or high-resolution CT in 424 individuals with PI*ZZ AATD genotype. 102 Instead, Wood et al., demonstrated that a single nucleotide polymorphism in the TNF-α gene (rs361525) was significantly associated with patients with AATD and chronic bronchitis (p=0.01). 102 SNPs in SERPINA1 may therefore not be the only genetic variants associated with AATD. An overview of the SNPs in SERPINA1 variants associated with AATD is further described by Foil 63 in a separate chapter of this review series.
Circulating free light chains (FLCs) of antibodies produced during an immune response are markers of adaptive immune activation and are clinically relevant biomarkers of several autoimmune diseases. 103 Increased FLC levels have also been reported in asthma and COPD.104,105 In AATD, patients that have chronically colonized sputum cultures have significantly higher FLC levels compared with patients with no positive cultures (p=0.008). 106 Furthermore, in severe AATD, increased FLC levels were significantly associated with mortality (p=0.001), indicating that FLCs could play a role in the risk-stratification of patients requiring more intensive monitoring and disease management. 106
MicroRNAs (miRNAs) primarily act as regulatory molecules for gene expression in normal physiology, but also play important roles in pathological processes in a wide variety of diseases, such as cancer, 107 cardiovascular disease, 108 rheumatic diseases, 109 and age-related diseases, such as osteoarthritis. 110 In a preliminary study, nine miRNAs were found to be significantly down-regulated in patients with AATD, which may provide the opportunity for miRNA-based monitoring of lung and liver disease associated with AATD. 111
Conclusions
Alternatives to IV AAT therapy with different modes of action are being investigated. In the short-term, use of chaperones could be a promising new treatment, providing that researchers can overcome the current requirement for large quantities of chaperones for an effective outcome. Use of in silico ligand screening may also prove to be a valuable tool in designing new molecules that interfere with AATD pathogenesis. In the long- term, hepatocyte replacement and CRISPR strategies would be greatly beneficial, as these methods would allow simultaneous treatment of both lung and liver disease associated with AATD. There are also new biomarkers being identified that may indicate levels of disease progression and response to therapy. However, it may be several years before any new therapies enter the market. At present, IV AAT therapy remains the only available disease-modifying and well tolerated pharmacological intervention for AATD.
Footnotes
Acknowledgements
Medical writing assistance was provided by Ben McDermott and Steven Foster of Meridian HealthComms Ltd., Plumley, UK, in accordance with good publication practice (GPP3), funded by CSL Behring.
Author contributions
FFR contributed to the writing of the manuscript, reviewed the manuscript, and approved the manuscript for submission.
Conflict of interest statement
FFR is a consultant, speaker, and researcher for Takeda and Grifols, and a consultant for CSL Behring.