
Abstract
Rasmussen’s encephalitis (RE) is rare neurological diseases characterized as epilepsia partialis continua, invariably hemiparesis, and cognitive impairment. This disease is encountered frequently in childhood and presents with progressive atrophy of the unilateral hemisphere, and there are also sustained neurological complications. Owing to uncertain pathogenesis, the most effective way to limit the influence of seizures currently is cerebral hemispherectomy. In this review, we focus on four main lines of pathogenesis: virus infection, antibody-mediated, cell-mediated immunity, and microglia activation. Although one or more antigenic epitopes may give rise to infiltrating T cell responses in RE brain tissue, no exact antigen was confirmed as the definite cause of the disease. On the other hand, the appearance of antibodies related with RE seem to be a secondary pathological process. Synthetic studies have suggested an adaptive immune mechanism mediated by CD8+ T cells and an innate immune mechanism mediated by activated microglia and neuroglia. Accordingly, opinions have been raised that immunomodulatory treatments aimed at initial damage to the brain that are induced by cytotoxic CD8+ T cell lymphocytes and microglia in the early stage of RE slow down disease progression. However, systematic exploration of the theory behind these therapeutic effects based on multicenter and large sample studies are needed. In addition, dysfunction of the adenosine system, including the main adenosine removing enzyme adenosine kinase and adenosine receptors, has been demonstrated in RE, which might provide a novel therapeutic target for treatment of RE in future.
Keywords
Introduction
Rasmussen’s encephalitis (RE), also known as Rasmussen syndrome, is a rare chronic disease of unknown origin, causing inflammation and creating a unilateral cerebral hemisphere lesion in the central nervous system (CNS). Theodore Rasmussen first described this devastating disease with its clinical and pathological characteristics in 1958. 1 Its clinical features are refractory epilepsy, and progressive neurological and cognitive impairment. The most characteristic representations are progress local atrophy of the cerebral cortex and epilepsia partialis continua (EPC). RE occurs especially in school-age children, imposing a huge family burden due to the inexorable atrophy of the cerebral hemisphere leading to further impairment of neurological function, and thus neuropsychological problems. Up to now, hemispherectomy is still the only efficient therapy. 2 In the past nearly 60 years, although we have made considerable advances in the study of this crippling disease, research into its pathogenesis, etiology and better methods of treatment is still ongoing. Consequently, it is necessary to extract research on pathogenesis and the direction of future treatment. In this review, we summarize the clinical features and recent fundamental research related to RE pathogenesis, looking forward to providing new insights for the treatment of RE.
Clinical features
Epidemiological research in Germany showed an estimated incidence rate of RE of about 2.4 patients per 10 million persons under 18 years old. 3 The mean age of onset in RE is 6 years, thus the disease occurs mainly in childhood or early adolescence, with no gender difference. 4 Diverse types of seizures, such as focal aware seizure and focal impaired awareness seizure, even associated secondary generalized tonic-clonic seizure, are a common clinical symptom of RE. EPC, which appear in half of cases, may occur with a rapid increase in seizure frequency, and is difficult to control with antiepileptic drugs (AEDs). The typical progression of RE is progressive unihemispheric cerebral neurological deficit within a few months of the initial seizure. Hemiplegia accompanies all stages of RE once it appears. Initially, movement deficiency is confined to the post-seizure phase, but then becomes serious and permanent as the disease continues to advance. If the lesion affects the dominant hemisphere, symptoms such as aphasia and hemianopia may occur. 5 RE patients reach a stable stage of neural function loss, called the residual stage, a few months to 1 year after origination.6,7 Cognitive impairment, another distinguishing feature of RE that is related to the severity of seizure, embodies mainly memory decline and lack of concentration and leads to learning disability. 8 In terms of the overall course of RE, three stages are proposed: (i) a prodromal stage with low frequency of seizure possibly accompanied by slight hemiplegia and an average duration of 7.1 months (0–8.1 years); (ii) an acute stage with frequent seizures for which AEDs are ineffective, especially in patients with EPC, and rapid neurological decline; (iii) a residual stage, progressive atrophy of the cerebral hemisphere, permanent neurological deficits, and persisting infrequent seizures as in the acute phase. 9
Abnormal electroencephalography (EEG) accompanies RE progression, although there is no specific EEG change to help make a definite diagnosis. In the initial stages of RE, EEG can manifest as slow background rhythms, with slow focal and epileptic abnormalities in the lesion hemisphere. During the subsequent few months, interictal abnormal discharge may appear independently in the unaffected hemisphere, which is indicative of the risk of overall cognitive decline rather than bilateral lesions of the brain (Figure 1K–L). 10 Neuroimaging examination provides significant evidence for the diagnosis and evaluation of RE. Magnetic resonance imaging (MRI) shows the unilateral hemispheric atrophy. The characteristic MRI features are the preferential atrophic process in the frontal lobe and the insula, enlargement of the lateral ventricle, atrophy of caudate nucleus, as well as hyperintensity of cortex and subcortical white matter on T2 and flair image. As the disease deteriorates, cortical atrophy often develops in the unilateral hemisphere (Figure 1A–J). Recently, it was proposed that gray matter in the contralesional hemisphere appeared to lose volume in subcortical gray matter structures; however, patient numbers in this study were limited. 11 In addition, automated MRI volumetric hemisphere analysis can be offered as an independent means of assessing RE progression. 12 Functional imaging, such as fluorodeoxyglucose-positron emission tomography (FDG-PET), reveals diffuse hypometabolism in the affected hemisphere. Moreover, single photon emission computed tomography (SPECT) demonstrates interictal hypoperfusion and ictal hyperperfusion of focal lesions. FDG-PET and SPECT to localize the RE increases diagnostic confidence, with especially effective detection of ictal onset area by ictal EEG in the absence of MRI abnormalities by the central benzodiazepine receptor (BZR)-SPECT.13,14

MRI imaging features of RE. This is a 5-year-old patient; image acquisition was at 17 months after seizure onset. (A–F) Left hemisphere atrophy in T1, T2, and T2 FLAIR image from axial and coronal scan. (G–H) PET showed left hemisphere hypometabolism. (I–J) T2 FLAIR image after surgical treatment of anatomic hemispherectomy. (K) EEG showing focal slow wave activity in the left hemisphere, which increased during the interictal period. (L) EEG showing EPC at right lower limbs in the ictal period.
At present, the clinical diagnosis of RE depends mainly on the European consensus criteria proposed in 2005. The diagnostic criteria are described two parts: Part A and Part B (Table 1). All three conditions in Part A or two conditions in Part B need to be satisfied for a diagnosis of RE. A study by Olson reported that the sensitivity of Bien’s diagnostic criteria is 81%, taking 82 patients with a differential diagnosis of RE in contrast to pathological examination as the diagnostic gold standard. 15 Although Bien’s diagnostic criteria have reliable sensitivity and specificity, Olson proposed that RE can also be diagnosed without evidence of EPC, progression of focal cortical deficits, or abnormal MRI in the case of positive pathological biopsy. In other words, B3 in Part B plus two conditions of Part A could also be used for diagnosis. 15 Moreover, Karla et al. suggested modified criteria, replacing “markedly lateralized” with “unihemispheric”, as a result of affecting contralateral hemisphere in abnormal EEG and cerebral atrophy of MRI as the disease progresses. 4 In our view, the evaluation criteria for diagnosis need a greater accumulation of clinical cases and adequate argument.
RE diagnostic criteria of Bien et al. in 2005. 2

Progressive means that at least two sequential clinical examinations or MRI studies are required to meet the respective criteria.
EEG, electroencephalography; EPC, epilepsia partialis continua; MRI, magnetic resonance imaging; RE, Rasmussen encephalitis.
The pathological peculiarities of RE are cortical inflammation activation, neuron loss, gliosis, microglial nodules, T-lymphocyte clusters, and perivascular cuffs in the unilateral hemisphere (Figure 2). According to these pathological characteristics, Robitaille et al. suggested that pathological changes in the cortex divide RE into four stages, from mild to severe. 16 The pathologic changes in the unihemisphere emerge with multifocal distribution at different stages, which corresponds with immune-mediated processing of neuronal loss. 17 A dual pathology has been reported in some cases, comprising cortical dysplasia, tuberous sclerosis, and ischaemic lesions.2,18–20 In recent years, the frequency of dual pathology, especially focal cortical dysplasia (FCD), seems to be on the rise.21–23 Further research into dual pathology may provide a new direction for the pathogenesis of RE.
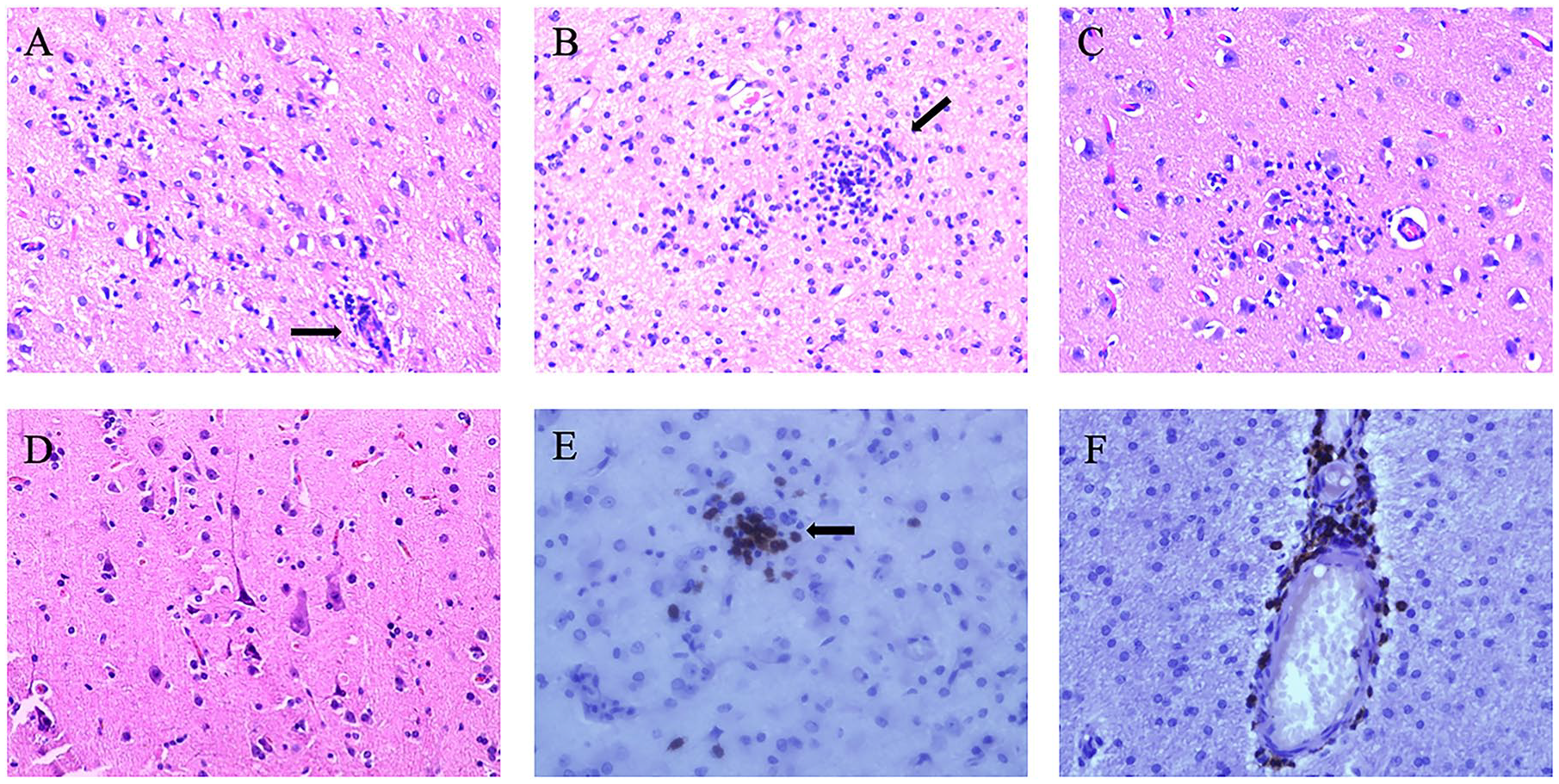
Characteristic neuropathological changes of RE. (A) Perivascular lymphocytic cuffing (arrow, H&E staining). (B) Microglial nodule formation (arrow, H&E staining). (C) Neuron undergoing neuronophagia (H&E stain). (D) Neuronal degeneration in the cerebral cortex (H&E stain). (E) Parenchymal lymphocytic in the cerebral cortex (arrows, CD8 immunostaining). (F) Perivascular lymphocytic cuffing in the cerebral cortex (CD8 immunostaining).
Although the etiology of RE is not completely clear, the main treatment is aimed at symptoms and inflammation, to control epilepsy, and to prevent further deterioration of neurological function, which means treatment with AEDs, immune treatments and surgery. Of course, symptomatic treatments such as vagus nerve stimulation (VNS) and transcranial stimulation also are means of sporadic palliative therapy according to the literature.24,25 AED treatment has only a limited effect on seizures. Immunotherapy, which is used mainly in the early stage of the disease, can relieve seizures or prevent immune-mediated hemispheric damage; however, these treatments also have only slight effects. Surgical treatment is a more satisfactory therapeutic strategy for RE.26,27 There are three main types of surgical operation: anatomical hemispherectomy, functional hemispherectomy, and disconnection hemisphere.28–30 Moreover, anatomical hemispherectomy surgery is more effective in controlling epileptic seizures within acceptable complications compared with hemisphere disconnection surgery and functional hemispherectomy surgery, as shown by a series of 45 cases at our epilepsy center. 31 Perfectly curable RE without any complications continues to be our ultimate aim, which certainly still requires a search for new treatment plans from the perspective of pathogenesis.
Etiology and pathogenesis
Hypothesis of virus infection mechanism
By virtue of the similar pathological process of chronic inflammation, and based on our knowledge of encephalitis, investigators looked at viral infection initially as a main research direction in the field of RE etiology. In addition, the characteristics of being confined to one side of the cerebral hemisphere seemed to fit the slow progression of viral infections. Our epilepsy center suggested that expression of Epstein-Barr virus antigen, human cytomegalovirus antigen, human herpes virus 6 antigen, and human papilloma virus antigen was obvious in RE brain.32–35 However, virus infection mechanisms could not illustrate the pathogenesis adequately and no study has detected virus replication in brain tissue of RE patients up to now. The reasons underlying virus antigen expression in RE need further investigation.
Antibody-mediated pathogenesis
The hypothesis of antibody-mediated pathogenesis of RE originated in 1994, following presentation of the histopathologic features of RE in two rabbits immunized with glutamate receptor (GluR3) after enhancing antibodies to recombinant GluRs. But what is more interesting is that anti-GluR3 antibody, via plasma exchange, efficiently improved seizure frequency and neurological deficits in one RE patient.
36
Results of other studies maintained that the reason for neuron loss in RE is related to an antibody-mediated response or the direct activation of ion channel receptors.37,38 Therefore, in the study of the pathogenesis of RE, antibody-mediated mechanisms currently occupy the dominant position. However, only a proportion of patients benefit from plasmapheresis therapy in a short period, whereas others show no improvement in clinical symptoms.39,40 Later research suggested that anti-GluR3 antibodies were detected in other types of epilepsy patients, and other anti-neuronal antibodies, such as Munc-18 and the alpha7-acetylcholine receptor, were identified in sera from a few patients with RE.41–43 Moreover, patients with limbic encephalitis, where seizure is the main clinical feature, had detectable leucin-rich glioma inactivated 1 (LGI1), α-amino-3-hydroxy-5-methyl-4-isoxazoleproprionic acid (AMPA) or gamma-aminobutyric acid (GABA) receptors antibodies, and were treated effectively by immunotherapy.44,45 Another prospective study suggested that a fraction of AED-resistant epilepsy patients presented neural autoantibodies, particularly antibodies binding to synaptic antigens.
46
Although one case reported that clinical syndrome and neuroimaging manifestations of anti-N-methyl-
Cell-mediated immunity mechanism
Recent studies have concentrated mainly on the function of cytotoxic T cells in the pathogenesis of RE. Pathologic examination of affected cerebral hemisphere revealed that cytotoxic CD8+ T cell lymphocytes occupy the majority of infiltrated T cells, and, more importantly, that, in the inflammatory lesions, about 10% of them are granzyme-B positive CD8+ T cells. This is viewed as a strong evidence of neuronal damage caused by cytotoxic T cells, as the proportion of granzyme-B+ T lymphocytes that gravitate toward the neuronal membrane exhibit features of polarization of cytotoxic granules. 48 Astrocytic degeneration, caused by cytotoxic T lymphocyte attack, gives rise to neuronal cell death and seizure induction. 49 Spectratyping of T cells in peripheral blood and corresponding brain specimens found that CD8+ T cell clones expanded with brain-restricted T-cell receptor (TCR) clonotypes, and proved an antigen-driven major histocompatibility complex (MHC)-I restricted immune response. 50 As CD8+ T cell expansion in peripheral blood was related to severity of RE disease, a probable way to improve symptoms was to restrict these cells infiltrating into the brain. 51 This evidence seemed to support the view that the T cell immune response in the brain was induced by particular antigens. The identity of these antigens, and whether they are intrinsic autoantigens or foreign antigens, is difficult to determine currently. Nevertheless, exemplary studies confirmed that non-MHC-restricted immune response also participated in pathogenesis of RE, on account of verifying γδ T cells with uniform TCR δ1 chain CDR3 sequences. 52 Indeed, CD4+ expanded T-cell clones and CD103+ resident memory T cells illustrate the complexity of brain-infiltrating T-cell immunity in RE.53,54
On the other hand, the idea that inflammatory cytokines are secreted from T cells has attracted increasing attention as it could be evidence of a cellular immune response. Research has shown that mRNA expression of interferon-γ (INF- γ) and five chemokines, including CCL5, CCL22, CCL23, CXCL9 and CXCL10, increased in RE brain tissue compared with brain specimens of cortical dysplasia. There is a marked negative correlation between the expression of these cytokines (IFN-γ, CXCL5, CXCL9, CXCL10) and the period from the onset of seizure to surgery. 55 CXCL10, which is expressed on neurons and astrocytes in surgical specimens of children with RE, is one of the known ligands of CXCR3, which is expressed on cytotoxic T lymphocytes. The CXCR3–CXCL10 axis plays a significant role in T cell recruitment into the affected hemisphere. 56 The existence of IFN-γ and tumor necrosis factor (TNF) secreted by CD4+ T cells was a supplement to previous studies. 54 Although the above studies suggested the view that the main pathogenesis of RE was an adaptive immune response, a general plan for immunomodulatory and immunosuppressive drug therapy needs more investigation.
Microglia-activation-mediated neurodegeneration
Microglia act as the resident innate immune cells in brain, and their activation state is one of most characteristic pathological hallmarks in RE. Activation levels of microglia in various regions of the brain are diverse, and are correlated with the infiltration of T cells, especially in the early pathological stage with cortical damage.17,57 So far, there are actually no details about the function of microglia emerging in the early stage of pathogenesis. With more research, the release by microglia of cytokines such as IL-1β and inflammasome activation was confirmed as inducing seizures and causing neuronal damage in other epileptic disorders.58–61 As the cause of cellular hyperexcitability, increased pannexin hemichannels linked to microglial activation manifested as epileptogenic mechanisms in RE. 62 Recent research has revealed that upregulated endosomal Toll-like receptor (TLRs) in microglia at pathological stage 0 provide a microenvironment for infiltration of T cells and attacking neurons at pathological stage 1, leading to even more activation of inflammasomes and chemokines. 63 This supplied direct evidence for microglia-activation-mediated neurodegeneration.
More and more attention has been paid to the activation of astrocytes in RE because this process is dominant at each stage of cortical damage, and is simultaneous with T lymphocyte infiltration and microglial activation.49,64 Because astrocytes participate in induction and perpetuation of the inflammatory response during the process of epileptogenesis, it is speculated that activated astrocytes are involved in the epileptogenic process of RE.65,66 Our resent work detected endogenous high-mobility group box-1 (HMGB1) binding to TLRs in the cytoplasm of activated astrocytes in RE specimens, which hinted that a HMGB1-TLR pro-inflammation pathway in astrocytes might represent a potential target. 67 The facility of astrocytes and microglia activation needs further study.
Future therapeutic directions: experimental and clinical evidence
Adenosine system dysfunction in RE
Adenosine – an inhibitory and protective modulator in the CNS – plays an active role in anticonvulsant and anti-inflammatory effects through regulating the balance of A1 receptor (A1R) and A2A receptor (A2AR).68–70 Moreover, the finding that overexpression of adenosine kinase (ADK) related with astrogliosis results in downregulated adenosine has been confirmed as being involved in epileptogenesis in both a rat epileptic model and epileptic human brain tissues.71–74 Focal adenosine augmentation therapy, in which treatment with ADK inhibitors corrected adenosine dysfunction, is well authenticated to reduce abnormal discharge of neurons and seizures.75,76 In our recent work on RE, excessive ADK accompanied by astrocytosis was examined in lesion cortical specimens using western blot analysis and immunohistochemistry. 77 At the same time, A1R positioned at the perinuclear region of neurons was also upregulated in the focus tissues. 78 Furthermore, our study demonstrated that upregulated A2AR led to downregulation of glutamate transporter GLT-1, and that neuron apoptosis was finally induced. 79 The above results proved that dysfunction of the adenosine system plays a significant role in the pathogenesis of RE, in particular in neuronal damage.
Mechanism of transporter-mediated GABA release in RE
The disruptive balance between GABA and glutamate is one of the key considerations in seizure. Chronic impairment of the GABAergic system has been proved in the epileptic cerebral cortex of human and animal models, resulting in a decrease in the seizure threshold.80,81 Enhancing GABA release by reducing extracellular calcium concentration ([Ca2+]e) is an effective mechanism against seizures, even as a potential drug treatment of EPC by compensating GABAergic interneuron dysfunction. 82 However, unlike the pathogenesis of other epilepsy, GABA release depending on calcium-withdrawal is decreased in RE. This supports a point of view in which Na+/Ca2+ exchanger activators could enhance GABA-transporter-mediated GABA release, so as to achieve the purpose of anti-seizure in early stage RE patients.83,84 Of importance, the further mechanism of this pathway remains to be studied.
Immunosuppressive or immunomodulatory treatment
In recent years, immunotherapy for RE had focused on infiltrative immunocytes and secreted cytokines described in some case reports. With its direct effect against B cells, Rituximab – a chimeric mouse-human monoclonal anti-CD20 antibody – reportedly led to one RE patient being seizure-free for several months. 85 Tacrolimus – a comprehensive inhibitor of T lymphocytes through limiting IL-2 – was applied as a treatment strategy after a few months of steroid pulse in the early stages of childhood RE. 86 Another randomized prospective study proposed that tacrolimus may prevent neurological deficit and development of refractory epilepsy. 3 In a case report, one patient diagnosed with RE selected treatment with mycophenolate mofetil, which leads to inhibition of lymphocyte proliferation after no response to corticosteroids and immunoglobulin treatment. 87 It is likely an effective treatment would need to impede inflammatory cytokines secreted from T cells and microglia. An open pilot study illustrated this point; adalimumab, as an immunoglobulin G1 (IgG1) monoclonal antibody acting on TNF-α, reduced the frequency of seizure and neurological impairment. 88 Yet adalimumab improved symptoms only in the early stage of RE and any curative effect needs further confirmation. 89 Another recent research study into the effectiveness of different immunotherapies reported that drugs targeting T-cells, such as cyclophosphamide, natalizumab and alemtuzumab, but not azathioprine, could reduce the inflammatory reaction of RE patients. 90 At present, there is more and more research into RE immunotherapy, with most studies focusing on the positive effectiveness of treatments, causing failed attempts to be neglected. Despite research into various immunotherapeutic strategies, therapeutic effects required to be verified through multicenter and large sample studies.
Conclusion
RE is rare progressive disease leading to disastrous EPC and neurological disorder. Hemispherectomy is considered to be the most direct cure for intractable epilepsy, but is unfortunately accompanied by the postoperative complications of hemiparesis and hemianopia. As the etiology of this disease is unclear, there is no specific therapeutic strategy aimed at pathogenic factors. Most work in the pathogenic area is devoted to the activation of infiltrating CD8+ T cells and microglia in order to explore better non-surgical treatments. Currently, immunotherapy, including tacrolimus and adalimumab, appears to be effective in the early stage of the disease in some cases. However, because the features of this disease are only gradually becoming clear, comprehensive judgments of early diagnosis have a certain degree of difficulty. In addition, developing study fields in RE, such as the adenosine system, bring a whole new direction to treatment possibilities. With such deep research into pathogenesis from experimental to clinical studies, the therapeutic dilemma posed by RE will be overcome in the foreseeable future.
Footnotes
Conflict of interest statement
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This project was supported by the National Natural Science Foundation of China (81571275).