
Abstract
Mitochondrial dysfunction is a common pathological hallmark in various inflammatory and degenerative diseases of the central nervous system, including multiple sclerosis (MS). We previously showed that oxidative stress alters axonal mitochondria, limiting their transport and inducing conformational changes that lead to axonal damage. Teriflunomide (TFN), an oral immunomodulatory drug approved for the treatment of relapsing forms of MS, reversibly inhibits dihydroorotate dehydrogenase (DHODH). DHODH is crucial for de novo pyrimidine biosynthesis and is the only mitochondrial enzyme in this pathway, thus conferring a link between inflammation, mitochondrial activity and axonal integrity.
Here, we investigated how DHODH inhibition may affect mitochondrial behavior in the context of oxidative stress. We employed a model of transected murine spinal roots, previously developed in our laboratory. Using confocal live imaging of axonal mitochondria, we showed that in unmanipulated axons, TFN increased significantly the mitochondria length without altering their transport features. In mitochondria challenged with 50 µM hydrogen peroxide (H2O2) to induce oxidative stress, the presence of TFN at 1 µM concentration was able to restore mitochondrial shape, motility, as well as mitochondrial oxidation potential to control levels. No effects were observed at 5 µM TFN, while some shape and motility parameters were restored to control levels at 50 µM TFN.
Thus, our data demonstrate an undescribed link between DHODH and mitochondrial dynamics and point to a potential neuroprotective effect of DHODH inhibition in the context of oxidative stress-induced damage of axonal mitochondria.
Keywords
Introduction
Multiple sclerosis (MS) is a chronic inflammatory disease of central nervous system (CNS) that affects more than 2.5 million people worldwide. 1 In MS, inflammation, demyelination and neurodegeneration are considered to contribute to disease development.2,3 It is assumed that in MS, a misguided immune response against the CNS is initiated and orchestrated by autoreactive T cells, leading to progressive demyelination, oligodendrocyte injury and axonal loss,4,5 that affect not only the white but also the grey matter. 6
The mechanisms by which neuroinflammation and myelin damage lead to neurodegeneration have not been fully elucidated; however, the sustained release of reactive oxygen species (ROS) and nitrogen species (NOS) by macrophages and activated microglia during inflammation appears to contribute to the damaging cascade.7–9 Also in cortical lesions, demyelination appears to be associated with excessive oxidative damage. 10 Mitochondrial pathology and subsequent focal axonal injury appears also to be triggered by inflammation-associated ROS and NOS and to be independent of demyelinating processes. 11 Axonal mitochondrial damage is an early sign of neurodegeneration that precedes and contributes to focal and reversible alterations in axon morphology. The alterations of the mitochondrial function within axons have been proposed to occur in the early stages of the disease, even before demyelination,11,12 and to precede neuronal death.13–16 In autopsied tissue from chronic progressive MS, respiratory deficient neurons were detected both in white and grey matter. Respiration deficits were shown to be caused by multiple deletions of mitochondrial DNA, probably subsequent to inflammation and oxidative stress, that contributed to an enhanced susceptibility of axons and neurons to additional damaging insults. 17 In this line, we and others have shown that oxidative stress disrupts the transport of mitochondria in the axon.14,18 Hence, mitochondrial dysfunction is considered as one of the major contributors of neuroaxonal damage in MS.
With regard to MS management and treat-ment, teriflunomide (TFN) (Aubagio; Genzyme, Cambridge, MA, USA) is a once-daily oral immunomodulatory drug for the treatment of patients with relapsing forms of MS. 19 TFN has been shown to reduce relapse events and increase the periods of remission.20,21
TFN seems to exert its therapeutic effect by non-competitively and reversibly inhibiting the mitochondrial respiratory chain-associated enzyme dihydroorotate dehydrogenase (DHODH).22–25 DHODH is involved in de novo pyrimidine biosynthesis, thus limiting lymphocytic proliferation and inflammation. However, whether the inhibition of DHODH alone may affect neuronal mitochondria remains uncertain. Importantly, a very recent retrospective, single-center, observational study indicated that the effect of TFN in reducing cortical grey matter atrophy is superior to the effect of the anti-oxidative and anti-inflammatory dimethyl fumarate. 26 Moreover, it has been reported that TFN penetrates into the CNS and exerts its effect directly within the brain. 27 Thus, TFN may indeed have the potential to affect axonal mitochondria directly.
To explore the possible effects of TFN on the nervous system, we have used in this study a previously established model of explanted spinal roots, in which we had shown that mitochondria undergo a series of alterations in response to oxidative stress.18,28
In patients treated daily with 14 mg TFN, average steady-state maximum TFN concentration (Cmax) in plasma is 168 μM. 29 The half maximum concentration (IC50) for interaction of TFN with human DHODH is 1 µM30,31 and 50–100 µM is considered sufficient to inhibit protein tyrosine kinase in vitro.29,31 Moreover, a study assessing the effect of TFN on eryptosis indicated that concentrations ranging from 3.7 to 37 µM TFN might compensate oxidative stress-mediated erythrocyte changes in vitro. 32 In rats, it has been shown that after one single injection of 10 µg/g TFN, approximately 2–4% of the blood concentration was found in the brain (~2.5–4.1 µM). 30 Although an extrapolation to the human reality is not exact, we could suppose in treated patients a TFN concentration within the nervous system of about 3–7 µM. Therefore, in our study, we investigated the effect of TFN on oxidative stress-induced mitochondrial alterations in murine root explants using three different TFN concentrations, 1 µM, 5 µM and 50 µM.
We show that TFN is able to prevent mitochondrial alterations induced by hydrogen peroxide (H2O2), suggesting that TFN has additional therapeutically relevant properties related to mitochondrial protection in axons. 31
Materials and methods
Ethics
All experimental procedures were approved by the local authority on animal studies in Berlin (Landesamt für Gesundheit und Soziales Berlin; ID: T0002/10). Animal studies were performed in strict accordance with the European Communities Council Directive of 22 September 2010 (2010/63/EU).
Solutions and drugs
Explanted roots were bathed in artificial cerebrospinal fluid (aCSF) containing the following: solution I: 124 mM sodium chloride (NaCl), 1.25 mM sodium dihydrogen phosphate (NaH2PO4), 10.0 mM glucose, 1.80 mM magnesium sulphate (MgSO4), 1.60 mM calcium chloride (CaCl2), 3.00 mM potassium chloride (KCl); solution II: 26.0 mM sodium bicarbonate (NaHCO3). Solutions I and II were mixed immediately before use. Hydrogen peroxide (H2O2; 30% w/w in H2O, with a stabilizer) and dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich. To induce oxidative stress, explanted roots were incubated with 50 µM of H2O2 dissolved in aCSF for 30 min. TFN was applied at different concentrations along with H2O2 for 30 min. TFN was provided in powder form by the manufacturer (Sanofi Genzyme), which was dissolved in DMSO and stored at −20°C.
Preparation of ventral spinal roots
Ventral spinal roots were prepared as described in our previous work.18,28,33 Briefly, C57BL/6 mice at least 3 weeks of age were anesthetized with isoflurane prior to cervical dislocation. After separating the connective tissue and exposing the dorsal side of the spinal cord, an initial sectioning was made at the thoracic level, which proceeded in a rostral to caudal direction until the last vertebrae. The spinal cord was lifted gently to expose the ventral roots, which were cut distal to the spinal cord but before the formation of the peripheral nerves. The explanted spinal cord with attached roots was transferred to aCSF saturated with carbogen (95% oxygen [O2]; 5% carbon dioxide [CO2]). Under a dissecting microscope, lumbar ventral roots of at least 0.8 cm were selected and separated from the spinal cord.
Labeling of mitochondria
All experimental incubations were conducted in a submerged incubation chamber (Brain Slice Keeper- BSK6-6; Scientific Systems Design Inc., Ontario, Canada), which allows for multiple treatment conditions and continuous carbogen perfusion of each submersion well. Transected ventral spinal roots were transferred to fresh aCSF containing 300 nM, MitoTracker CMTMRos orange (Life Technologies, Darmstadt, Germany) for 30 min and washed with aCSF.
Confocal microscopy
Explanted ventral roots were placed onto a glass coverslip and transferred to an imaging chamber filled with carbogenated aCSF. To prevent movement of the roots during imaging, a custom-built net was placed on the top of the roots. 28 For all imaging experiments, we used an inverted laser-scanning confocal microscope adapted for live cell imaging (LSM 710; Carl Zeiss, Jena, Germany). MitoTracker Orange was excited with a diode-pumped solid state (DPSS) laser at 561 nm. After finding the middle of the root, 3 × 60-sec videos (2 sec/frame) with a resolution of 512 × 512 pixels were acquired in three separate regions of interest (ROI) according to the following criteria: (a) there was a clearly defined node of Ranvier; (b) there were visibly labeled mitochondria; and (c) the areas were no closer than 0.2 cm from the ends of the roots. Typically, the first ROI was located at the middle of the root, whereas the following two were to the right and left of the middle region.
Analysis of mitochondrial dynamics
Mitochondrial morphology was assessed using an automated analysis function of the Volocity 6.3 software (Perkin Elmer, Rodgau, Germany). The first frame of every video was used for analysis. Shape factor, a measure of circularity ranging from 0 and 1 (closer to ‘0’ was a longer mitochondrion, whereas ‘1’ was a perfect circle), length (µm) and area of individual mitochondria were quantified for assessing change in mitochondrial morphology.
Mitochondrial transport was quantified in terms of the number of moving mitochondria, velocity, displacement, and track length of moving mitochondria. Displacement is the measure of shortest distance in µm, covered by a mitochondrion; it was measured as a straight line from the start to the end position during the 30 frames. Track length is the measure of real distance path longitude followed by the mitochondrion. Mitochondria were tracked manually using Volocity 6.3 software (Perkin Elmer, Rodgau, Germany). Any mitochondrion with a displacement of at least 1 µm was considered ‘mobile’. Measurements from three ROI were averaged for each root.
In total, 15 different mice in 15 independent experiments were investigated. Depending on the quality of the explants, at least five independent roots and 15 ROI per culturing condition were included into the analysis (usually up to three different ROI per root). Specifically, 39 and 44 ROI were analyzed for the untreated group and H2O2 treated groups, respectively; 15–21 ROI were used to investigate treatments with H2O2 + TFN. For the morphological investigations, the number of mitochondria in the selected ROI were 2586, 2860, 1306, 875 and 1550 for untreated, H2O2-treated, H2O2 + TFN (1 µM), H2O2 + TFN (5 µM) and H2O2 + TFN (50 µM), respectively. Analyses of motility included 201, 70, 83, 52 and 64 motile mitochondria for each of the above-mentioned groups.
For the comparison of untreated versus TFN-treated nerves, four independent experiments were performed. Analyses include 12 ROI per condition. In total, 568 and 672 individual mitochondria were analyzed, respectively.
Quantification of relative change in intracellular ROS
The fluorescence intensity of the MitoTracker Orange was quantified as a measure of intracellular ROS as described by Kweon et al., 34 The images obtained from confocal microscopy were used for the quantification of mitochondrial fluorescence intensity using Image J software.
Statistical analysis and data representation
The data were analyzed with Prism 5.01 software (GraphPad, CA, USA). All datasets were subjected first to D’Agostino and Pearson omnibus K2 normality test for Gaussian distribution. All data fitting the criteria for a normal distribution were subsequently analyzed using a one-way analysis of variance (ANOVA) with Bonferroni’s post hoc test. All data following a non-parametric distribution were analyzed using a Kruskal–Wallis test followed by a post hoc Dunn’s multiple comparisons test. All data are given in mean ± SD.
Data are shown in Tukey box and whisker plots. The box and whisker plot shows simultaneously the minimum, first quartile, mean (+), median (dissecting line inside the box), third quartile, and maximum of the data set. Whiskers indicate variability outside the upper and lower quartiles. Outliers are plotted as individual dots that are in line with whiskers. The mean + SD values are given in the corresponding tables.
Results
TFN altered mitochondrial dynamics in peripheral root explants
We labelled peripheral root mitochondria with MitoTracker Orange and the explants were imaged for morphological investigation (Figure 1a, b). Then, the effects of TFN on mitochondrial morphology and transport in unmanipulated explanted roots were investigated. Explanted roots were incubated in the presence or absence of 50 µM TFN. TFN treatment resulted in a statistically significant decrease in mitochondrial circularity (Figure 1c) and an increase in mitochondrial length (Figure 1d). There were no significant changes in mitochondrial area (Figure 1e) after TFN treatment compared with the untreated controls.

Teriflunomide (TFN) affected mitochondrial shape and length in untreated root explants.
For mitochondrial motility, TFN did not significantly change the number of motile mitochondria (Figure 2a) as well as the distance covered by the mitochondria (Figure 2c, d). However, it induced a significant reduction of the mean velocity of mitochondrial transport (Figure 2b). Corresponding statistical information is summarized in Table 1.

Teriflunomide (TFN) reduced mitochondrial velocity without influencing motile number, trajectory length and displacement of mitochondria in untreated root explants.
Summary of morphology and motility parameters of mitochondria in untreated and teriflunomide-treated peripheral root explants.

Values are shown as mean ± SD.
p < 0.05; **p < 0.01
TFN prevented oxidative stress-induced morphological changes in mitochondria
We previously reported that oxidative stress leads to substantial changes to both morphology and transport of axonal mitochondria. 18 Here, we investigated whether TFN, applied together with H2O2, would be able to prevent these effects. We treated the roots with 50 µM H2O2 (both groups containing the vehicle DMSO), and 3 different concentrations of TFN: 1, 5 and 50 µM in aCSF (Figure 3a–e). We analyzed a total of 39 untreated ROI, 44 ROI treated with H2O2, and 18, 15 and 21 ROI treated with 1 µM, 5 µM and 50 µM TFN in the presence of 50 µM H2O2, respectively, from 15 independent experiments. Consistent with our previous findings, we observed that treatment with 50 µM H2O2 induced an overall increase of mitochondrial circularity and a corresponding decrease in mitochondrial length and area. In particular, mitochondria were significantly more circular (Figure 3f), shorter (Figure 3g) and smaller (Figure 3h) than their untreated counterparts.

Mitochondrial morphology altered during oxidative stress with/out teriflunomide (TFN) treatment.
In the presence of 1 µM TFN, the shape factor of the mitochondria was reduced, that is, mitochondria became elongated or rod-shaped (Figure 3f). In contrast, no effects were observed at higher concentration of TFN. Moreover, the lowest and highest TFN concentrations (1 µM and 50 µM) induced a significant increase in mitochondrial length (Figure 3g) and area (Figure 3h), in comparison with the mitochondria exposed to H2O2 alone. Paradoxically, treatment with 5 µM TFN with 50 µM H2O2, showed no statistically significant effect on H2O2-induced morphological alterations (Figure 3f–h) (0.59 ± 0.19 shape factor, 1.43 ± 1.29 µm length, and 0.51 ± 0.69 µm2 area; Kruskal-Wallis test followed by Dunn’s post hoc test p > 0.1 in all cases).
Corresponding statistical information is summarized in Table 2.
Summary of shape factor, length and area of mitochondria under H2O2 treatment alone, and with 50 µM H2O2 in the presence of 1 µM, 5 µM and 50 µM teriflunomide.

KW, Kruskal–Wallis; H2O2, hydrogen peroxide.
Values are shown as mean ± SD.
p < 0.05; **p < 0.01; ***p < 0.001.
TFN prevented oxidative stress-induced changes in mitochondrial motility
To investigate TFN effects on mitochondrial motility, roots were treated either with aCSF, 50 µM H2O2 (both groups containing DMSO) or 50 µM H2O2 in the presence of three different concentrations of TFN: 1, 5 and 50 µM (Figure 4a–e). We observed that H2O2 treatment led to an overall decrease in the number of motile mitochondria (Figure 4f). In addition, the moving mitochondria had lower mean velocity (Figure 4g), trajectory length (Figure 4h), and displacement (Figure 4i) than the untreated mitochondria.

Mitochondrial motility altered during oxidative stress with or without teriflunomide (TFN) treatment. A representative image of mitochondrial tracking in (a) untreated, (b) hydrogen peroxide (H2O2)-treated, and (c, d, and e) H2O2-TFN treated where TFN was 1, 5 and 50 µM, respectively, in murine peripheral root explants. (f) Number, (g) velocity, (h) trajectory, and (i) displacement of mitochondria.
Again, the lowest and highest TFN concentration (1 µM and 50 µM) restored the motility-related parameters to control levels, except for the mitochondrial velocity with 50 µM TFN, when compared with the mitochondria exposed to H2O2 alone; for the number of moving mitochondria (Figure 4f), velocity (Figure 4g), trajectory (Figure 4h), and displacement (Figure 4i). In contrast, 5 µM TFN had no effect. Corresponding statistical information is summarized in Table 3.
Summary of mitochondrial motility, velocity, displacement and trajectory length under H2O2 treatment alone, and along with different concentrations of teriflunomide.

KW, Kruskal–Wallis; H2O2, hydrogen peroxide.
Values are shown as mean ± SD.
p < 0.05; **p < 0.01; ***p < 0.001.
TFN prevented change in mitochondrial oxidation potential in peripheral nerve explants during oxidative stress
MitoTracker Orange CMTMRos is a reduced, non-fluorescent dye that fluoresces on oxidation. Thus, in conditions of high oxidative stress, mitochondria acquire higher fluorescence intensity. 34 We observed that fluorescence intensity was higher in H2O2-treated roots compared with untreated controls (Figure 5). In the presence of 1 µM TFN, the fluorescence intensity in the mitochondria was reduced, approaching the values of the untreated axons (Table 4), suggesting that the H2O2-mediated increase in the oxidation potential could be prevented by TFN. In contrast, no effect was observed at 5 or 50 µM TFN. Corresponding statistical information is summarized in Table 4.
Summary of mean fluorescence intensities of mitochondria.
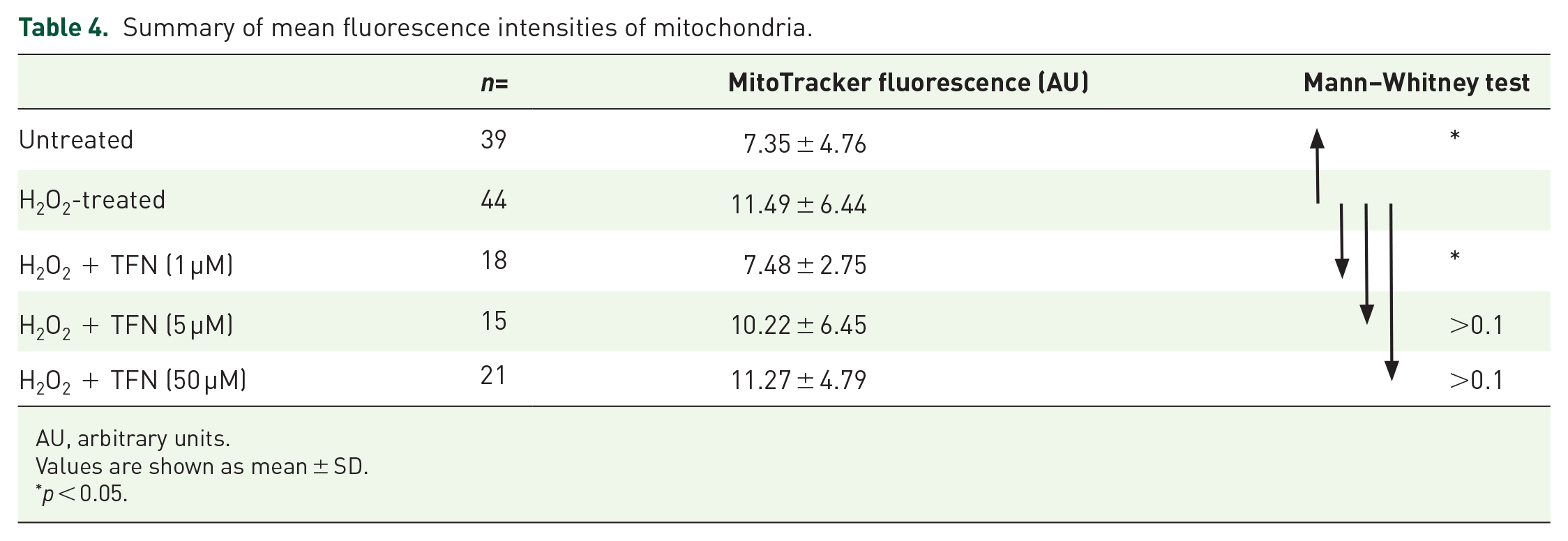
AU, arbitrary units.
Values are shown as mean ± SD.
p < 0.05.
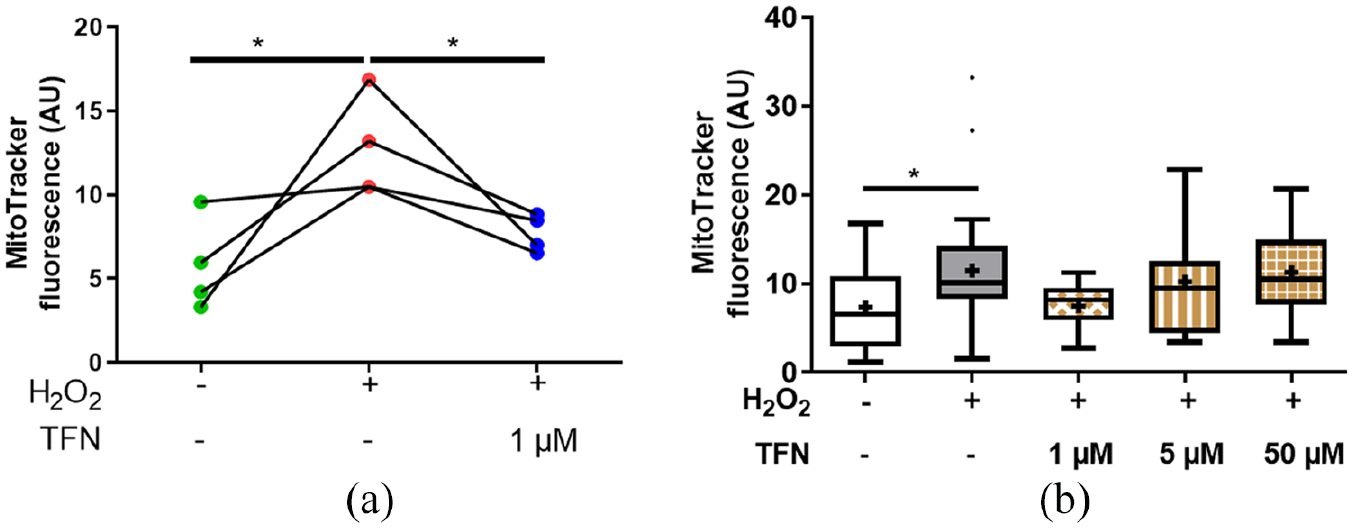
Teriflunomide (TFN) at 1 µM altered the oxidation potential of mitochondria, reducing oxidative MitoTracker Orange fluorescence intensity Fluorescence intensity of MitoTracker Orange staining depicting significant reduction of oxidation potential of 1 µM TFN. (b) MitoTracker Orange fluorescence intensity at 1, 5 and 50 µM TFN treatment during oxidative stress in comparison with untreated and H2O2-treated roots.
Discussion
While current treatments for MS focus on reducing inflammation via modulation of the immune system,25,35 there is a general lack of treatment targeting inflammation-promoted neurodegeneration,36,37 which is an integral component of disability progression.37,38 As early mitochondrial alterations are reported in inflammatory neurodegenerative diseases,8,11–16 the maintenance of mitochondrial integrity could be a key goal to achieve neuronal protection during neuroinflammation. 39 Here, we hypothesized that TFN, due to its ability to inhibit DHODH, 40 an enzyme functionally linked with complex III activity of the mitochondrial respiratory chain, 41 may influence mitochondrial stability in the context of oxidative stress. To test our hypothesis, we used our previously established model of murine explanted ventral roots, in which the morphology and the transport of mitochondria can be analyzed within peripheral axons.18,28
After excluding any relevant effect of DMSO (the dilution vehicle), in both untreated and H2O2-treated mitochondria (Supplemental Figures 1–2), we investigated the effect of TFN treatment in unmanipulated or oxidative stress-exposed spinal root explants.
Interestingly, in non-stressed roots, TFN seems to promote mitochondrial fusion, induce mitochondrial elongation (Figure 1d, e), and reduce mitochondrial velocity (Figure 2b). Mitochondrial fusion is important for the formation of mitochondrial networking that assists in reshuffling and redistributing the mitochondrial content.42,43 Thus, the inhibition of DHODH and subsequent effects on complex III of the electron transport chain (ETC) and respiration 41 may promote mitochondrial fusion as an attempt to redistribute the electron transport complexes that are still capable of maintaining the proton gradient and synthesizing adenosine triphosphate (ATP).
In contrast, during oxidative stress, we observed a reduction in mitochondrial length and size, which is indicative of fragmentation/fission of mitochondria that might undergo mitophagy. Mitochondrial fission has also been proposed to increase the number of mitochondria and their cellular distribution in order to meet the increasing energy demands of the cell.44,45 Although mitochondrial fission is extensively discussed in terms of mitophagy as well as apoptosis,43–46 intensive mitochondrial fission may translate into mitochondrial failure and the strategy to optimize mitochondrial functionality before undergoing apoptosis. It also serves to get rid of damaged, irreparable mitochondrial parts.45,47 Importantly, in the presence of 1 µM and 50 µM TFN, reduction in mitochondrial length and area due to oxidative insult could be prevented (Figure 3g, h). Intensive fragmentation during oxidative stress could not be prevented with 5 µM TFN.
Further, consistent with previous findings, we observed a reduced mitochondrial motility during oxidative stress,14,18 that is, reduced trajectories and transport velocity. The impairment of mitochondrial transport was preserved with TFN treatment. In axons, around 10–30% of mitochondria are motile, while more than 70% remain stationary. 48 This motile and stationary pool of mitochondria is dependent on the current energy demands of the cell.49,50 In addition, disrupted motility could lead to impairment of mitochondrial fusion.46,49,51 Thus, TFN may promote fusion by influencing the motility. On the other hand, it has been proposed that inhibition of DHODH by TFN may reduce the total amount of ROS in the cell. 52 Thus, TFN-mediated ROS reduction may also lead indirectly to an increased motility of stressed mitochondria.
Along this line, to assess the effects of TFN on ROS in our system, we monitored the fluorescence intensity of MitoTracker Orange CMTMRos (see Methods). 34 As expected, fluorescence intensity of CMTMRos significantly increased with H2O2 treatment, while inhibition of DHODH with 1 µM TFN reduced the ROS level (Figure 5). As complex III of the ETC is considered one of the major contributors to ROS formation, its compromised activity in the presence of TFN might reduce ROS production in mitochondria in peripheral spinal root explants. 5 µM and 50 µM TFN could not effectively reduce ROS, which might be attributed to the inhibition at higher concentrations of additional signaling pathways including tyrosine kinases. 53
On the other hand, the intermediate dose of 5 µM TFN showed no effects on H2O2-induced shape or motility changes. Why, in our experimental set-up a dose effect is missing, remains uncertain. The high variability of our data, which is intrinsic to the nature of mitochondrial dynamics and reflects the heterogeneity of the mitochondrial population in both physiological and diseased conditions, could have contributed to mask a true dose effect. To minimize this problem, several experiments with large amounts of mitochondria were analyzed (see Methods section). Moreover, depending on its concentration, TFN may function by a different mode of action. While low TFN concentrations are effective in inhibiting DHODH (1–1.5 µM), concentrations needed to achieve DHODH-independent effects such as inhibition of protein tyrosine kinase or cyclooxygenase-2 are much higher (50–200 μM). 29 However, little is known about the mode of action of intermediate concentrations. One could speculate that in our model, TFN at 5 µM may achieve partially known or yet undefined DHODH-independent effects that rather counterbalance the beneficial effects observed at 1 µM, while at 50 µM DHODH-dependent and independent mechanisms may synergize against dysfunctions observed under oxidative stress. Future experiments using long-living explants are needed to evaluate to what extent TFN effects at different concentration are DHODH-dependent and thus reversible.
Our previous data on root explants demonstrated that mitochondrial alterations caused by oxidative stress precede axonal damage. 18 Now, we show that these alterations could be pharmacologically reversed in vitro by TFN. Targeting dysfunction of axonal mitochondria should become one of the key goals in drug development not only for MS but also for other classic neurodegenerative disorders such as Parkinson’s or Alzheimer diseases. 54 In this line, we showed in the animal model of MS a protective effect of epigallocatechin-3-gallate (EGCG),55,56 a polyphenol, that among others, inhibits the formation of ROS and protects neurons.57,58 Also dimethyl fumarate, used to treat MS, prevents oxidative stress-related mitochondrial dysfunction, apoptosis and autophagy in murine oligodendrocytes in vitro. 59 Importantly, endogenous substances currently being investigated in MS may be exploited as therapeutics due their mito-protective capacities, such as high dose biotin, 60 vitamin D 61 or octadecaneuropeptide, a neurotrophic peptide produced principally by astrocytes, which is able to counteract oxidative stress-induced alterations. 62
In summary, our present findings suggest a protective effect of TFN on axonal mitochondria exposed to oxidative stress. Investigations expanding on these findings are needed to determine whether mitochondrial protection at the axonal level can be translated into protection of axons and neurons.
Supplemental Material
Malla_et_al._Supplementary_data – Supplemental material for Teriflunomide preserves peripheral nerve mitochondria from oxidative stress-mediated alterations
Supplemental material, Malla_et_al._Supplementary_data for Teriflunomide preserves peripheral nerve mitochondria from oxidative stress-mediated alterations by Bimala Malla, Samuel Cotten, Rebecca Ulshoefer, Friedemann Paul, Anja E. Hauser, Raluca Niesner, Helena Bros and Carmen Infante-Duarte in Therapeutic Advances in Chronic Disease
Footnotes
Conflict of interest
The authors declare that there is no conflict of interest.
Funding
The authors disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This work was funded by a research grant from Sanofi Genzyme to C.I-D. A.E.H, was supported by DFG TRR130, TP17. The authors also acknowledge support from the German Research Foundation (DFG) and the Open Access Publication Funds of Charité- Universitätsmedizin.
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.