
Abstract
Diabetic sarcopenia is a common complication of diabetes, substantially impacting patients’ quality of life and prognosis. Its pathogenesis is closely related to energy metabolism, with mitochondria—often referred to as the cellular “powerhouse”—playing a central role in this process. This review focuses on the role of mitochondrial dysfunction in diabetic sarcopenia, emphasizing mechanisms such as energy metabolism imbalance, oxidative stress–induced damage, and abnormalities in mitochondrial biogenesis and dynamics. Additionally, we discussed current research on diagnostic and therapeutic strategies targeting mitochondrial dysfunction. This narrative review aims to provide a theoretical foundation for a deeper understanding of the pathophysiology of diabetic sarcopenia and the development of novel therapeutic approaches.
Keywords
Introduction
Diabetes mellitus is a global health challenge, which affected 529 million people in 2021, with projections indicating an increase to 1.31 billion by 2050. China has the largest diabetic population worldwide, with over 118 million patients with diabetes, accounting for approximately 22% of the global diabetic population. 1 Sarcopenia is an age-related disorder characterized by progressive loss of muscle mass and function, substantially impacting overall survival and increasing the risk of postoperative complications. 2
A growing body of research has demonstrated a close association between sarcopenia and diabetes. Hyperglycemia can induce sarcopenia through multiple mechanisms, including disruption of the insulin signaling pathway, inhibition of protein synthesis, impairment of muscle microcirculation, promotion of oxidative stress, mitochondrial dysfunction, and abnormalities in mitochondrial dynamics. 3 Among these mechanisms, mitochondrial dysfunction plays a central role, as mitochondria are the key organelles responsible for cellular energy production and their functional integrity is crucial for maintaining muscle structure and function. The coexistence of diabetes mellitus and sarcopenia synergistically exacerbates insulin resistance and oxidative stress, leading to an accelerated decline in muscle mass and strength, which considerably increases the risks of functional disability, hospitalization, cardiovascular events, and mortality. Therefore, a comprehensive exploration of mitochondrial dysfunction in diabetic sarcopenia, along with effective diagnostic approaches and potential therapeutic strategies, holds significant scientific and clinical value for the prevention and treatment of diabetic sarcopenia, ultimately contributing to improved public health outcomes. Adhering to the Scale for the Assessment of Narrative Review Articles (SANRA) guidelines, 4 this narrative review focuses on type 2 diabetic sarcopenia, aiming to explore the bidirectional crosstalk between mitochondrial dysfunction and insulin resistance and propose targeted therapeutic interventions.
Overview of diabetic sarcopenia
Chronic hyperglycemia in diabetes induces a series of complex pathophysiological changes. Persistent hyperglycemia can lead to neurovascular complications, compromising neural trophic support to muscles and reducing vascular perfusion to muscle tissues. The European Working Group on Sarcopenia in Older People defines sarcopenia as a decline in skeletal muscle size, mass, and strength, leading to reduced walking speed, shorter walking distances, and decreased handgrip strength, which ultimately affect patients’ quality of life. 5
Sarcopenia can be classified into two major types: primary and secondary. Primary sarcopenia is commonly observed in the older population as a consequence of the natural aging process, resulting in irreversible and progressive loss of muscle mass. In contrast, secondary sarcopenia is associated with underlying diseases such as malignancies, malnutrition, and diabetes, with diabetes being a particularly significant contributing factor. 6
Insulin resistance is a key pathological mechanism of type 2 diabetes mellitus (T2DM). Beyond its role in glucose metabolism, insulin resistance negatively affects muscle protein synthesis. Under physiological conditions, insulin promotes amino acid uptake by muscle cells, thereby facilitating protein synthesis. However, in the presence of insulin resistance, this protein synthesis pathway is impaired, whereas the ubiquitin–proteasome system and calpain-mediated proteolysis are abnormally activated, accelerating muscle protein degradation. 7 Over time, the imbalance between reduced protein synthesis and increased protein degradation results in progressive muscle dysfunction and atrophy. The coexistence of diabetes mellitus and sarcopenia represents a critical clinical intersection with bidirectional pathophysiological interplay. An epidemiological study 2 revealed that patients with diabetes exhibit a 2- to 3-fold higher prevalence of sarcopenia than nondiabetic populations, which is attributable to shared mechanisms such as insulin resistance, oxidative stress, and chronic low-grade inflammation. This synergy accelerates muscle proteolysis and impairs mitochondrial function, leading to a rapid decline in muscle mass (8%–25% per decade after the age of 40 years) and strength, compounded by diabetes-related microvascular damage and lipid infiltration in muscle tissues. Clinically, this dual pathology substantially elevates functional disability risks (e.g. mobility loss and falls), hospitalization rates, and cardiovascular mortality while also exacerbating diabetic complications such as peripheral neuropathy and coronary disease. Early screening for sarcopenia in middle-aged patients with diabetes, particularly through modalities such as imaging technology and functional indicators, is imperative, as timely interventions (e.g. exercise therapy and glycemic control) may reverse muscle deterioration and improve long-term outcomes.
Overview of mitochondrial dysfunction
Mitochondria play a central role in cellular activities and are often referred to as the “powerhouse” of the cell. Through oxidative phosphorylation, mitochondria convert the energy released from the oxidative breakdown of nutrients such as glucose and fatty acids into adenosine triphosphate (ATP), the primary energy currency of cells. During oxidative phosphorylation, the electron transport chain (ETC) complexes are crucial for energy conversion. Under normal mitochondrial function, electrons are transferred sequentially along the ETC complexes, accompanied with proton translocation across the mitochondrial membrane, establishing the mitochondrial membrane potential, which drives ATP synthesis. However, when mitochondrial dysfunction occurs, this intricate energy production process is disrupted. Furthermore, there is a decline in the activity of ETC complexes, leading to impaired electron transfer, reduction in mitochondrial membrane potential, and subsequent decrease in ATP production. 8
During energy metabolism, mitochondria inevitably generate reactive oxygen species (ROS). Under physiological conditions, intracellular antioxidant systems efficiently neutralize ROS to maintain cellular homeostasis. However, mitochondrial dysfunction results in excessive ROS production, overwhelming the antioxidant defense mechanisms and inducing oxidative stress. 9 Oxidative stress, in turn, damages key mitochondrial macromolecules such as mitochondrial DNA (mtDNA), membrane proteins, and lipids, leading to mtDNA mutations, protein oxidative modifications, and lipid peroxidation. These detrimental effects contribute to mitochondrial injury. In addition, mitochondrial biogenesis is impaired, indicating that the synthesis of new mitochondria fails to meet cellular demands, resulting in a relative reduction in mitochondrial quantity. In terms of mitochondrial dynamics, the balance between fusion and fission is disrupted, leading to structural disorganization of the mitochondrial network. These changes further weaken mitochondrial function, ultimately impairing cellular energy supply and overall metabolic balance. 10
Disruptions in energy metabolism are a fundamental pathophysiological mechanism of sarcopenia, and mitochondria play a critical role in this process. Although mitochondrial content and activity naturally decline with aging, regular physical exercise can help maintain skeletal muscle energy metabolism and slow the progression of sarcopenia. In sarcopenia, the expression of genes associated with mitochondrial dysfunction is upregulated. 11 A genetic analysis of individuals from different ethnic backgrounds revealed that mitochondrial dysfunction significantly contributes to age-related degenerative changes in skeletal muscle mass and function. 12 Another study demonstrated that mitochondrial dysfunction leads to the excessive depletion of four mitochondrial-specific monolysocardiolipins, a pathway that may also contribute to the onset of sarcopenia. 13
Mechanisms of mitochondrial dysfunction in diabetic sarcopenia
Impairment of the insulin signaling pathway
As a fundamental pathological mechanism of diabetes, insulin resistance plays a critical role in diabetic sarcopenia, and mitochondrial dysfunction is intricately associated with the insulin signaling pathway. In diabetic sarcopenia, mitochondrial dysfunction alters cellular energy status, leading to reduced ATP production and an increased adenosine monophosphate (AMP)/ATP ratio. This change activates AMP-activated protein kinase (AMPK), which exerts dual effects on insulin signaling. On one hand, AMPK phosphorylates and activates insulin receptor (IR) substrate (IRS) proteins, facilitating downstream insulin signal transduction. On the other hand, AMPK indirectly modulates insulin-related proteins by downregulating the expression of mammalian target of rapamycin (mTOR), inhibiting its negative feedback regulation on IRS, thereby improving insulin sensitivity. This enhances glucose uptake and utilization in muscle cells, supplying more energy to muscles while also promoting muscle protein synthesis and storage. 14
However, mitochondrial dysfunction also leads to excessive production of ROS, inducing oxidative stress, which is closely associated with insulin resistance. Oxidative stress disrupts insulin signaling and induces insulin resistance by interfering with adipokine regulation. Specifically, oxidative stress activates several serine/threonine kinase pathways, such as IKKβ/nuclear factor-kappa B (NF-κB) and c-Jun N-terminal kinase (JNK), which phosphorylate IRS proteins and promote their degradation. Excessive ROS further impair insulin signal transduction by inhibiting GLUT-4 translocation to the plasma membrane. 15 Additionally, oxidative stress reduces protein kinase B (PKB) phosphorylation, decreases serine phosphorylation at IRS-1 Ser307, and downregulates GLUT-4 expression, all of which contribute to insulin resistance. Furthermore, oxidative stress interacts with inflammatory responses, damaging IR function and exacerbating insulin resistance. 16
Impairment of muscle oxygen supply and protein metabolism
Healthy mitochondria regulate vascular homeostasis by producing vasoactive factors such as vascular endothelial growth factor (VEGF), promoting angiogenesis, and maintaining endothelial cell function to ensure adequate blood perfusion to muscle tissues. 17 However, mitochondrial dysfunction reduces VEGF production, impairs endothelial cell function, decreases vasodilation capacity, and disrupts the structure and integrity of microvasculature, leading to microcirculatory dysfunction in muscles. Inadequate blood supply deprives muscle cells of sufficient oxygen and nutrients while impairing the clearance of metabolic waste, exacerbating muscle damage and dysfunction, thereby creating a vicious cycle. 18
Moreover, mitochondrial dysfunction results in ATP deficiency, inhibiting the mTOR signaling pathway, a key regulator of protein synthesis. Reduced mTOR activity directly leads to decreased protein synthesis. 19 Simultaneously, cellular stress responses induced by mitochondrial dysfunction, such as oxidative stress and endoplasmic reticulum stress, activate protein degradation pathways. For instance, the expression of E3 ubiquitin ligases, including muscle RING-finger protein-1 (MuRF1) and muscle atrophy F-box protein (atrogin-1), is upregulated. These proteins tag muscle proteins for degradation by the proteasome, accelerating muscle protein breakdown, disrupting the balance of muscle protein metabolism, and ultimately leading to progressive loss of muscle mass. 20
Oxidative stress and mitochondrial damage
Mitochondrial dysfunction is frequently accompanied with elevated oxidative stress. In diabetic sarcopenia, abnormalities in the mitochondrial ETC result in excessive ROS accumulation. Owing to their high reactivity, ROS inflict damage on various mitochondrial macromolecules. For example, ROS directly attack the phospholipid bilayer of the mitochondrial membrane, altering its fluidity and permeability. 21 Additionally, oxidative modifications of protein subunits in respiratory chain complexes reduce their enzymatic activity, further exacerbating mitochondrial dysfunction. 22
ROS-induced damage to mtDNA is particularly severe, as mtDNA lacks histone protection and has limited repair capacity. ROS-induced mtDNA mutations impair mitochondrial gene expression and disrupt the assembly of respiratory chain complexes, leading to further mitochondrial dysfunction.
Beyond direct mitochondrial damage, excessive ROS spread throughout muscle cells, causing widespread oxidative stress damage to various cellular components. At the protein level, ROS oxidize amino acid residues, altering protein structure and function, thereby disrupting intracellular signaling, enzymatic activity, and protein–protein interactions. 23 At the lipid level, ROS-induced lipid peroxidation compromises membrane integrity, affecting membrane fluidity and permeability, which interferes with cellular exchange and signal transduction. 24 At the nucleic acid level, ROS cause DNA strand breaks and base modifications, disrupting gene expression and impairing cell proliferation and differentiation. 25 The cumulative effects of oxidative stress ultimately lead to muscle cell dysfunction, increased apoptosis, and a gradual decline in muscle mass and strength.
Mitochondrial dysfunction is also a central driver of age-related sarcopenia, necessitating a clear distinction from diabetic sarcopenia. Diabetic sarcopenia is initiated by metabolic disturbances. Hyperglycemia and insulin resistance directly suppress mitochondrial oxidative phosphorylation by activating nicotinamide adenine dinucleotide phosphate (NADPH) oxidase and chronic inflammation (e.g. elevated interleukin (IL)-6 and tumor necrosis factor (TNF)-α levels), diabetes-specific microvascular damage induces muscle hypoxia, and lipid infiltration induces mitochondrial lipotoxicity, collectively accelerating Drp1-mediated excessive mitochondrial fission and energy metabolism collapse.
In contrast, age-related sarcopenia primarily stems from an age-related decline in mitochondrial quality control. Specifically, accumulated mitochondrial DNA mutations and impaired mitophagy lead to damaged mitochondrial accumulation, coupled with Mfn2 downregulation-induced fusion defects. Additionally, aging-associated hormonal changes (e.g. reduced growth hormone) exacerbate protein synthesis inhibition. Together, these processes contribute to progressive loss of muscle mass and function. 26
Clinically, diabetic sarcopenia exhibits higher muscle loss rates (up to 2%–3% annually) due to synergistic metabolic–inflammatory–vascular insults, which mutually exacerbate diabetic complications (e.g. cardiovascular disease and neuropathy) and markedly elevate mortality. Age-related sarcopenia, however, manifests as a decline in muscle function commensurate with aging, albeit progressing more gradually.
Energy metabolism imbalance
Mitochondrial dysfunction directly disrupts cellular energy metabolism. Under normal conditions, muscle cells rely on mitochondrial oxidative phosphorylation to generate ATP, meeting the energy demands of muscle contraction and other physiological activities. However, in diabetes, hyperglycemia and insulin resistance impair oxidative phosphorylation, leading to ETC dysfunction, suppression of aerobic respiration, and significant reduction in ATP production. As muscle contraction depends on an adequate ATP supply, energy deficiency weakens muscle contractility, ultimately leading to muscle atrophy and functional decline.
To sustain essential cellular activities, muscle cells compensate by shifting their energy metabolism strategy, increasingly relying on anaerobic glycolysis. Although anaerobic glycolysis provides a rapid but limited ATP supply, it is far less efficient than aerobic respiration and results in the accumulation of metabolic byproducts such as lactate. Excess lactate lowers muscle tissue pH, affecting enzymatic activity and further impairing muscle energy metabolism and function. 27 Moreover, energy metabolism imbalance disrupts intracellular ion homeostasis, particularly calcium homeostasis, which negatively impacts muscle contraction and relaxation, further exacerbating muscle weakness in patients with diabetic sarcopenia.
Mitochondrial biogenesis and dynamics abnormalities
Mitochondrial dynamics maintains network homeostasis through fusion and fission processes, regulated by core proteins including the fission protein Drp1 and fusion proteins Mfn1/2 and OPA1. 28 Drp1 mediates mitochondrial membrane constriction via GTPase activity-dependent mechanisms, requiring adaptor proteins (e.g. mitochondrial fission factor (MFF)) to localize to mitochondria–endoplasmic reticulum contact sites. Mfn1/2 achieves outer membrane fusion through transdimerization, whereas OPA1 preserves the inner membrane cristae architecture. Under imbalanced conditions (e.g. Drp1 hyperactivation or Mfn1/2 deficiency), exacerbated mitochondrial fragmentation leads to energy metabolism dysfunction and quality control failure.
In diabetic myopathy, hyperglycemia and oxidative stress enhance the mitochondrial translocation of Drp1 by suppressing its Ser637 phosphorylation, resulting in excessive fission. Concurrently, marked downregulation of Mfn2 expression impairs mitochondria–endoplasmic reticulum interaction, inducing calcium overload and insulin resistance. The Drp1/Mfn1/2 imbalance further reduces mitochondrial fusion, generating fragmented networks that suppress ATP production and accelerate muscle atrophy.
Excessive ROS generated by mitochondrial fission activates NF-κB and NLRP3 inflammasome, promoting the release of inflammatory cytokines including IL-6 and IL-1β. Inflammatory signaling (e.g. IL-6–Janus kinase/signal transducer and activator of transcription (JAK/STAT) pathway) reciprocally inhibits mitochondrial biogenesis master regulator peroxisome proliferator-activated receptor gamma coactivator-1 alpha (PGC-1α) while enhancing Drp1 activity, establishing a “ROS inflammation–mitochondrial damage” positive feedback loop that persistently exacerbates muscle degeneration.
The NLRP3 inflammasome is activated by mitochondrial ROS and leaked mtDNA, driving caspase-1-mediated cleavage of pro-IL-1β to release mature IL-1β that directly impairs mitochondrial membrane potential. IL-6 aggravates mitochondrial oxidative damage by suppressing Sirt3 deacetylase activity and reducing SOD2 antioxidant defense. Furthermore, NLRP3 activation promotes the expression of OMA1, the OPA1 cleavage enzyme, which inhibits inner membrane fusion and causes cristae structure disintegration, ultimately leading to complete mitochondrial functional collapse.
Relationship between mitophagy, ferroptosis, and sarcopenia
Mitophagy and ferroptosis exhibit synergistic destructive effects in the pathogenesis of diabetic sarcopenia. 29 Dysfunctional mitophagy leads to the accumulation of damaged mitochondria, inducing ROS overproduction and metabolic imbalance, which directly impair skeletal muscle cells. These excessive ROS further activate iron-dependent lipid peroxidation, inducing ferroptosis that causes sarcolemmal disintegration and impairs satellite cell–mediated muscle regeneration, thereby establishing a vicious cycle of “mitochondrial damage–ferroptosis activation.” This interplay involves three core mechanisms: (a) imbalance in mitochondrial dynamics proteins (Drp1/Mfn1/2); (b) aberrant ferritinophagy mediated by the iron metabolism regulator NCOA4; and (c) persistent activation of the NLRP3 inflammasome. Targeting these pathways through combined interventions—such as exercise therapy to enhance mitophagy coupled with iron chelators to suppress ferroptosis—may disrupt this pathological cascade, offering novel therapeutic strategies to ameliorate muscle atrophy.
Mechanistic interplay between mitochondrial dysfunction and insulin signaling pathways
Mitochondrial dysfunction and insulin signaling pathways engage in a pathological crosstalk central to diabetes progression. 30 Hyperglycemia and oxidative stress induce mitochondrial damage via NADPH oxidase activation and chronic inflammation (e.g. elevated IL-6/TNF-α levels), suppressing oxidative phosphorylation (OXPHOS) and reducing ATP production. Insufficient ATP impairs IR-mediated PI3K/Akt signaling, inhibiting GLUT-4 membrane translocation and glucose uptake, while mitochondrial ROS exacerbate insulin resistance by promoting IRS-1 serine phosphorylation (e.g. Ser307) via JNK/SAPK and p38 mitogen-activated protein kinase (MAPK) pathways. This establishes a self-perpetuating “oxidative stress–mitochondrial damage–insulin resistance” cycle. At the molecular level, hyperglycemia disrupts mitochondrial dynamics by overactivating the fission protein Drp1 and suppressing the fusion protein Mfn2, driving mitochondrial fragmentation and metabolic collapse. Concurrently, impaired Akt/mTOR signaling due to ATP depletion undermines protein synthesis and cell survival. Disease progression reveals stage-specific dynamics: (a) early compensatory β-cell hyperinsulinemia coincides with mitochondrial ROS accumulation; (b) the progressive phase features mitochondrial DNA mutations, mitophagy decline, and sustained downregulation of insulin signaling components (GLUT-4 and IRS-1); and (c) late-stage decompensation involves β-cell apoptosis (mitochondria-dependent) and irreversible insulin resistance in muscle/adipose tissue. These interactions highlight the mitochondria–insulin axis as a therapeutic nexus for disrupting diabetic pathophysiology.
Figure 1 schematically demonstrates the bidirectional pathogenic network linking mitochondrial dysfunction and insulin resistance.
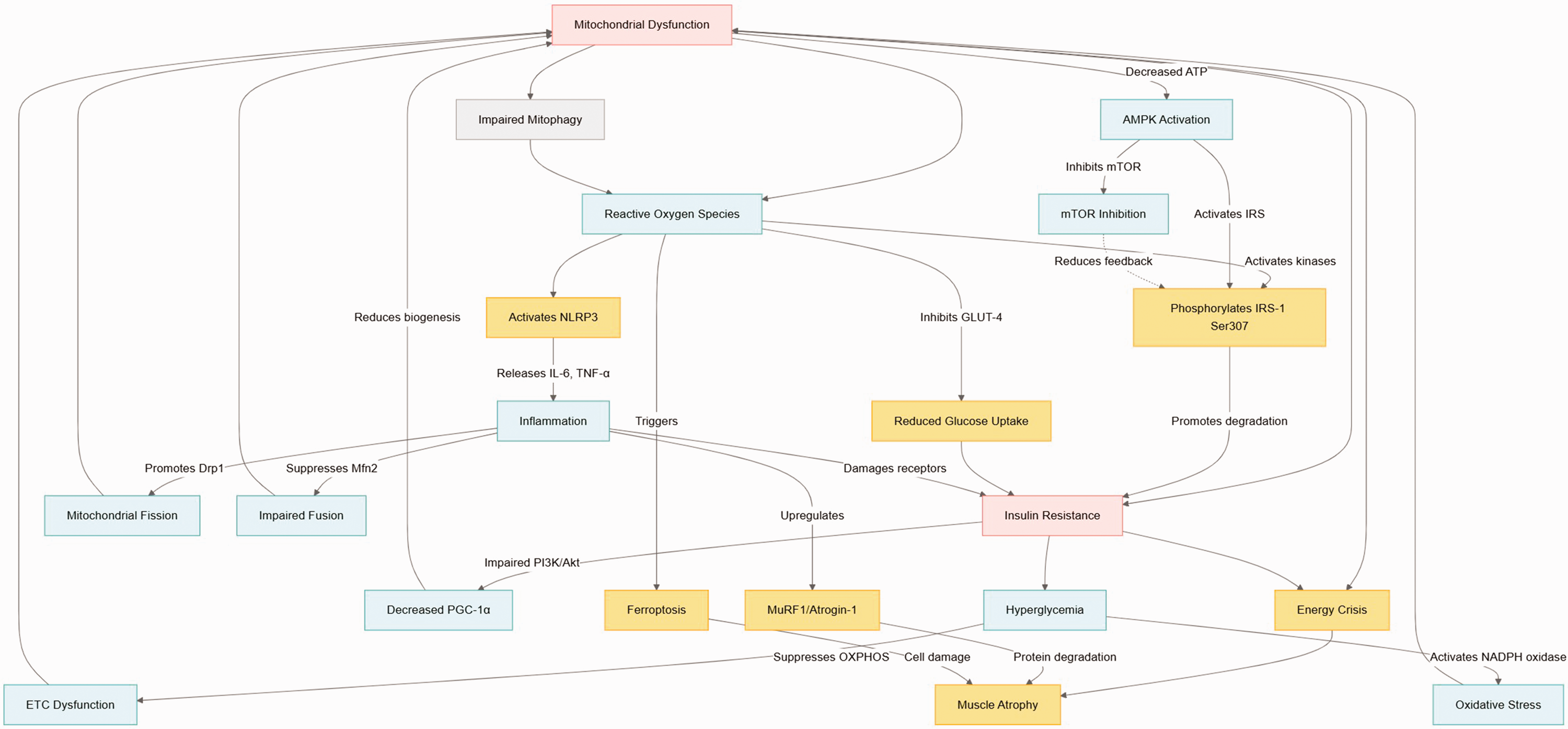
Mitochondria–insulin resistance interplay.
Diagnosis of diabetic sarcopenia
Imaging techniques
Computed tomography (CT) and magnetic resonance imaging are considered the gold standard for assessing muscle and fat mass owing to their precision in differentiating adipose tissue from other soft tissues. However, their high cost and radiation exposure (particularly with CT) limit widespread clinical use. Dual-energy X-ray absorptiometry offers a cost-effective, low-radiation alternative for quantifying whole-body or regional lean and fat mass, making it the recommended modality in routine practice.
In recent years, muscle ultrasonography has emerged as a potential diagnostic tool for sarcopenia. Current investigations have demonstrated that the combined use of the cross-sectional area and echo intensity parameters, when applied to the rectus femoris or biceps brachii muscles, demonstrates superior diagnostic accuracy compared with individual parameters. 31 Future efforts should prioritize the standardization of measurement protocols and validation of novel ultrasound modalities, such as elastography, to enhance diagnostic reliability. 32
Functional indicators
Functional indicators primarily comprise muscle strength and mass. Muscle strength assessment recommends grip strength dynamometry, with cutoff values referenced to geographically specific norms. This is because diminished grip strength demonstrates a significant correlation with adverse clinical outcomes. Functional testing combines the chair stand test (>15 s for five repetitions) with timed-up and go (≥20 s), gait speed (≤0.8 m/s), or short physical performance battery score (≤8 points) to evaluate global muscle function and coordination, where abnormalities indicate “probable sarcopenia.”
Muscle mass evaluation prioritizes the measurement of body mass index (BMI)–adjusted calf circumference (<33 cm in males, <32 cm in females; adjustments of ±4–12 cm based on BMI stratification), with abnormal findings necessitating confirmation via imaging modalities. Diagnostic synthesis mandates a multimodal framework integrating functional deficits with quantifiable muscle loss to establish a definitive diagnosis.
Therapeutic strategies targeting mitochondrial dysfunction in diabetic sarcopenia
Treatment strategies for diabetic sarcopenia primarily include nutritional interventions, pharmacological treatments, and exercise therapy (Figure 2). Studies have shown that selective androgen receptor modulators, antidiabetic drugs, and renin–angiotensin system inhibitors can mitigate age-related muscle deterioration while enhancing muscle strength and mass. 33 The key molecular targets for treating diabetic sarcopenia include insulin-like growth factor-1, protein kinase B (Akt), mTOR, transforming growth factor-beta, and NF-κB/MAPK signaling pathways. 34
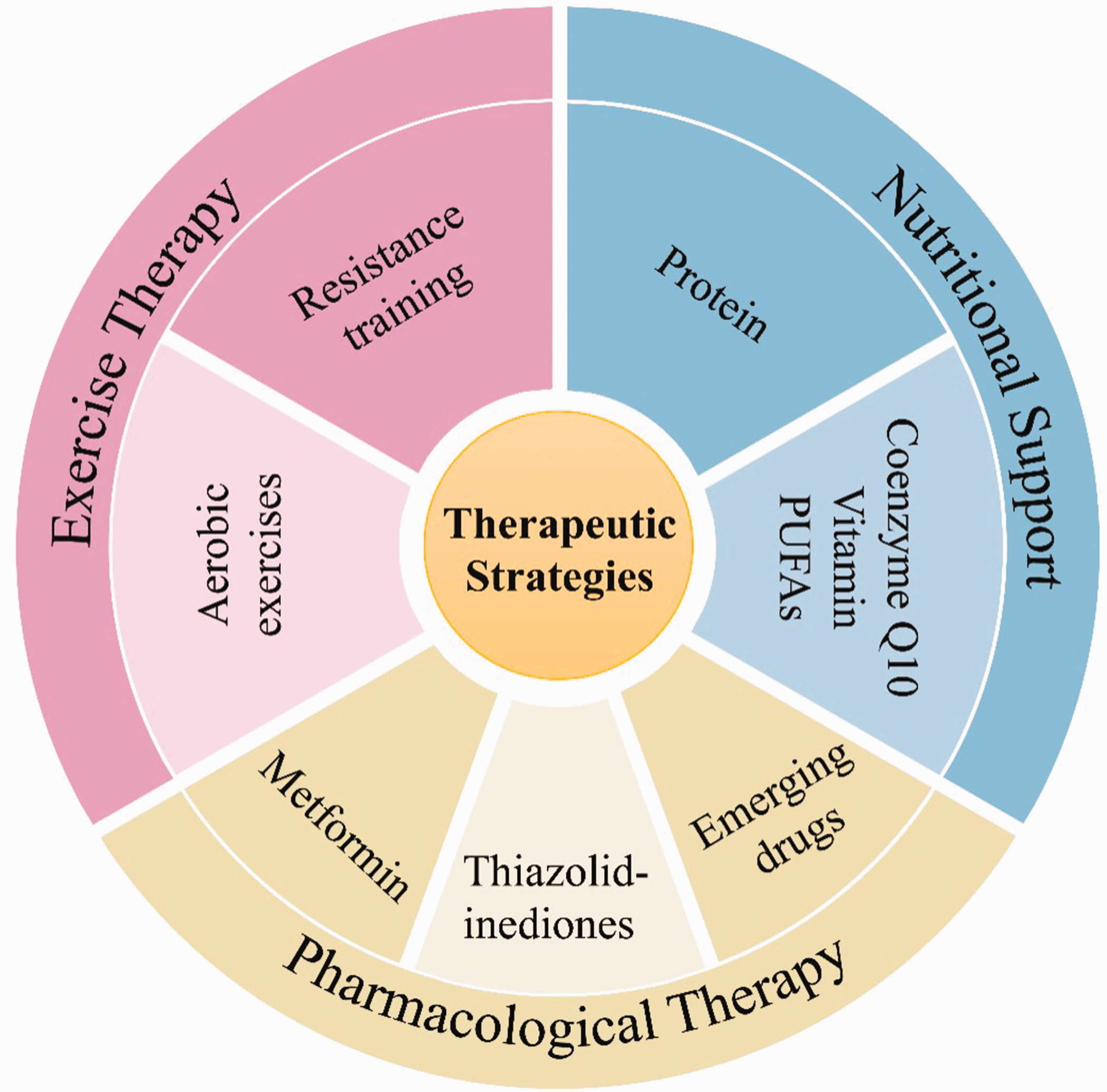
Therapeutic strategies targeting mitochondrial dysfunction in diabetic sarcopenia.
Exercise therapy
Exercise therapy is one of the most effective strategies for improving mitochondrial dysfunction in patients with diabetic sarcopenia. Aerobic exercises such as brisk walking, jogging, and swimming substantially enhance mitochondrial respiratory function. During exercise, the energy demands of muscle cells increase substantially, prompting mitochondria to accelerate electron transport and oxidative phosphorylation, thereby improving the activity of respiratory chain complexes. 35 Long-term aerobic exercise also upregulates the expression of PGC-1α, a key gene involved in mitochondrial biogenesis, promoting the synthesis of new mitochondria and increasing the number and volume of mitochondria in muscle cells. 36
Resistance training, including anaerobic exercises such as weightlifting, push-ups, and squats, primarily focuses on enhancing muscle strength. Muscle contraction and relaxation during resistance training activate intracellular signaling pathways, such as the mTOR pathway, which promotes muscle protein synthesis while indirectly improving mitochondrial function. 37 Additionally, resistance training increases capillary density in muscle tissue, improving microcirculation and ensuring a sufficient supply of oxygen and nutrients to mitochondria, thereby further optimizing mitochondrial function and alleviating muscle weakness and atrophy in patients with diabetic sarcopenia. 38
Progressive resistance training (PRT) is considered one of the most effective interventions for early-stage sarcopenia. 39 A randomized controlled trial demonstrated that PRT improves gait speed, muscle strength, and body composition in sarcopenic individuals. 39 However, the study lacked data on the effects of exercise on metabolic and inflammatory markers in patients with sarcopenic obesity.
A meta-analysis by 37 Pan et al. 40 included studies involving 2208 patients with T2DM. In this meta-analysis, eight exercise therapies, namely, supervised aerobic exercise, unsupervised aerobic exercise, anaerobic exercise, supervised resistance exercise, unsupervised resistance exercise, combined exercise, flexibility exercise, and no exercise, were compared to evaluate the effects of different exercise modalities on improving glycemic control, weight loss, and cardiovascular risk factors in patients with T2DM. The findings demonstrated that combined exercise methods yielded optimal improvements in hemoglobin A1c (HbA1c) levels. Therefore, this should be considered when prescribing exercise regimens, 41 with exercise prescriptions incorporating simultaneous aerobic, anaerobic, resistance, and flexibility training rather than being limited to a single modality.
Nutritional support
Adequate nutritional support is essential for improving mitochondrial dysfunction in diabetic sarcopenia. Protein is a fundamental component of muscle, and sufficient protein intake is crucial for maintaining muscle mass and promoting mitochondrial biogenesis. High-quality protein sources such as lean meats, fish, eggs, and legumes provide essential amino acids necessary for muscle protein synthesis while also supporting protein synthesis during mitochondrial biogenesis. 42
Additionally, specific nutrients play important roles in the regulation of mitochondrial function. Coenzyme Q10, a critical cofactor in the mitochondrial ETC, enhances mitochondrial respiration, improves ATP production efficiency, and mitigates oxidative stress–induced mitochondrial damage. 43 Antioxidant vitamins such as vitamin C, vitamin E, and β-carotene help neutralize intracellular ROS and reduce oxidative damage to mitochondria. Furthermore, omega-3 polyunsaturated fatty acids, including eicosapentaenoic acid and docosahexaenoic acid found in fish oil, improve mitochondrial membrane fluidity and permeability, stabilize mitochondrial function, and optimize energy metabolism, thus serving as a potential adjunctive therapy for diabetic sarcopenia. 44
Pharmacological therapy
Metformin, a first-line treatment for T2DM, not only lowers blood glucose levels but also positively influences mitochondrial function. Metformin activates AMPK, which promotes mitochondrial biogenesis through the AMPK signaling pathway, thereby increasing mitochondrial quantity and function. Additionally, AMPK activation enhances insulin sensitivity, regulates muscle protein metabolism, and reduces muscle protein degradation. 45
Thiazolidinediones such as rosiglitazone modulate peroxisome proliferator-activated receptor gamma (PPARγ) activity, influencing the expression of mitochondrial-related genes, improving mitochondrial energy metabolism efficiency, and alleviating oxidative stress in mitochondria. 46 Certain natural compounds have also shown promise in ameliorating mitochondrial dysfunction. For instance, resveratrol exhibits antioxidant and anti-inflammatory properties and can activate SIRT1, which subsequently upregulates PGC-1α expression, promoting mitochondrial biogenesis while inhibiting oxidative stress and mitochondrial deterioration. This provides a potential therapeutic approach for diabetic sarcopenia.
Resveratrol exhibits distinct dose-dependent characteristics in terms of potential adverse effects and dosage limitations. 47 Clinical evidence 47 indicates that high-dose regimens (e.g. ≥1 g/day) may induce gastrointestinal disturbances (including diarrhea and nausea) and hepatorenal dysfunction. Notably, a phase II clinical trial (NCT00920556) reported that 5 g/day dosing caused renal toxicity and fatigue in patients with multiple myeloma, with fatal cases documented. Furthermore, resveratrol interferes with drug metabolism through cytochrome P450 inhibition (particularly CYP3A4), possibly potentiating the toxicity of anticoagulants or chemotherapeutic agents. Its low bioavailability (oral absorption <5%) necessitates high clinical doses; however, animal studies demonstrate that ≥1000 mg/kg doses can induce oxidative stress and organ damage. The current recommended safe dosage is 450 mg/day for a 60 kg adult, although precise dosing requires individualized assessment considering metabolic variability and concomitant medications.
Moreover, several emerging drugs are currently under development or are being tested in clinical trials, aiming to target mitochondrial dysfunction. Some compounds, such as SIRT1 agonists (e.g. SRT1720), enhance mitochondrial biogenesis by activating the PGC-1α signaling pathway. 48 Additionally, mitochondrial-targeted antioxidants such as mitoquinone (MitoQ) have shown promise in selectively accumulating within mitochondria, effectively scavenging ROS, reducing oxidative stress damage, and improving mitochondrial function, offering new hope for the pharmacological treatment of diabetic sarcopenia. 49 According to a research progress report by Shaito et al., 50 the potential adverse effects and dosage limitations of MitoQ exhibit significant concentration- and context-dependent characteristics. Clinical studies have demonstrated that high-dose regimens (e.g. 40–80 mg/day) may induce dose-dependent gastrointestinal reactions (including nausea and vomiting), with this dosage range established as the maximum tolerated dose in a 12-month trial involving patients with Parkinson’s disease (n = 128). Conversely, in acute pancreatitis models, 500 μM MitoQ was shown to impair mitochondrial respiratory function and induce acinar cell apoptosis. Animal studies further revealed that chronic administration of 10 mg/kg MitoQ leads to hepatic congestion in rats. Despite these risks, phase II clinical trials confirmed that the same dosage range (40–80 mg/day) effectively reduces liver injury in patients with hepatitis C. The current recommended dose is 10 mg/day (based on the manufacturer’s guidelines), although clinical evidence remains limited, necessitating cautious dose adjustments tailored to individual metabolic profiles.
Conclusion and future perspectives
Diabetic sarcopenia is a complex and challenging complication of diabetes, wherein mitochondrial dysfunction plays a central role in its pathogenesis. Impaired mitochondrial oxidative phosphorylation results in insufficient ATP production, compelling muscle cells to shift to an inefficient glycolytic energy supply. This metabolic shift is accompanied with lactic acid accumulation and ion homeostasis imbalance, ultimately weakening muscle contraction ability. Conversely, abnormal mitochondrial ETC induces a ROS burst. This ROS overproduction not only damages key components such as mtDNA and membrane proteins but also drives chronic inflammation by activating the NF-κB/NLRP3 pathway, thereby further inhibiting mitochondrial biogenesis. Additionally, excessive activation of Drp1 and defective expression of Mfn2 lead to mitochondrial fragmentation, disrupt the mitochondrial–endoplasmic reticulum calcium signal interaction, and exacerbate insulin resistance and protein metabolism disorders.
Current diagnostic approaches rely on the combined evaluation of functional indicators obtained from imaging techniques. Exercise therapy, nutritional support, and drug interventions demonstrate broad potential in treating diabetic sarcopenia by targeting mitochondrial dysfunction.
Notwithstanding the gradually increasing prevalence of sarcopenia among patients with diabetes, no specific targeted drugs have been developed to date, and our understanding of its pathogenesis remains insufficient. Developing drugs with higher specificity for the key targets in the pathogenesis of diabetic sarcopenia is of utmost importance. Furthermore, the interindividual differences in mitochondrial dysfunction and treatment response are not fully elucidated, and there is a dearth of precision medicine approaches tailored to diverse patient groups. Moreover, the long-term efficacy and safety of existing treatment modalities necessitate further clinical validation.
Future research endeavors should focus on elucidating the molecular mechanisms underlying mitochondrial dysfunction in diabetic sarcopenia. It is also essential to gain more in-depth insights into the long-term progression of type 2 diabetes complicated with sarcopenia through large-scale prospective cohort studies, identify more specific and effective therapeutic targets, and develop novel drugs and combination treatment strategies. Advancements in modern biotechnology, including gene editing and stem cell therapy, may offer innovative approaches for the treatment of diabetic sarcopenia, ultimately enhancing patient outcomes and reducing the socioeconomic burden associated with this condition.
Footnotes
Acknowledgements
AI tools were used to improve language clarity. All content decisions remain the authors’ responsibility.
Authors’ contributions
Conceptualization: Zhilin Xie, Xiaoliu Li, Jing Zhang
Methodology: Zhilin Xie, Jing Zhang
Formal analysis: Zhilin Xie, Xiaoliu Li
Investigation: Zhilin Xie, Yuanhao Li
Writing—original draft: Zhilin Xie, Yuanhao Li, Xiaoliu Li
Writing—review and editing: Zhilin Xie, Yuanhao Li, Jing Zhang
Funding: Jing Zhang, Xiaoliu Li
Project administration: Jing Zhang, Xiaoliu Li
Data availability statement
No new datasets were generated or analyzed in this narrative review. All data cited in this article are derived from publicly available sources, with complete references listed in the bibliography.
Declaration of conflicting interests
The authors declare no competing interests.
Funding
This study was supported by Hainan Provincial Natural Science Foundation of China (No. 821RC756). Mechanistic study of the autophagy pathway Omi/HtrA2 based on the study of electroacupuncture combined with exercise training in T2DM myasthenia gravis. Science and Technology Department of Hainan Province. The New Medical Technology Research and Transformation Seed Program of Shanghai Municipal Health Commission Study on multimodal rehabilitation detection and treatment of patients with hemiplegia based on portable intelligent devices (No.2024ZZ1030), and Shanghai Minhang District Medical Specialty Construction Project (No.2025MWFC04).