
Abstract
A consensus has formed based on epidemiological studies and clinical trials that intervention to reduce low density lipoprotein cholesterol (LDL-C) will reduce cardiovascular disease (CVD) events. This has progressively reduced the thresholds for intervention and targets for treatment. Whist statins are sufficient for many people in primary prevention, they only partially achieve the newer targets of secondary prevention for established CVD. Increasing use of statins has highlighted that 1–2% cannot tolerate these drugs. Other cholesterol-lowering drugs such as ezetimibe add to the benefits of statins but have limited efficacy. The discovery of activating mutations in proprotein convertase subtilisin kexin-9 (PCSK9) as a cause of familial hypercholesterolaemia while inactivating mutations lower LDL-C led to the idea to develop PCSK9 inhibitors as drugs. This article reviews the history of lipid-lowering therapies, the discovery of PCSK9 and the development of PCSK9 inhibitors. It reviews the key trials of the current antibody-based drugs and how these have influenced new guidelines. It also reviews the controversy caused by their cost and the increasing application of health economics to determine the optimum strategy for implementation of novel therapeutic pathways and surveys other options for targeting PCSK9 as well as other LDL-C lowering compounds in late development.
Keywords
Introduction
The history of intervention on low-density lipoprotein cholesterol
The role of lipids in cardiovascular disease (CVD) was established in epidemiological studies such as Framingham or the Munster Heart Study about 50 years ago, but its occurrence had been described in histological specimens of atheromatous plaque about 100 years ago by Anitschow.1–3 The role of lipids in atherosclerosis has been widely recognised and so the trials of lipid-lowering drugs in patients with moderate elevations of low-density lipoprotein cholesterol (LDL-C) have a long history. In hyperlipidaemia (as opposed to hypertension) genetic causes of elevated lipids are frequent. Familial hypercholesterolaemia (FH) caused by defects affecting the function of the LDL receptor pathway and presenting consequently with elevated LDL-C and premature CVD has an incidence of 1 in 350. 4 Unfortunately, there have been no CVD outcomes studies in FH with any lipid-lowering therapy despite its frequency. The contrast with trials in type 1 diabetes is notable.
Initial attempts at intervention in the 1970s with drugs such as niacin, bile acid sequestrants and clofibrate in patients with moderate hypercholesterolaemia (with or without CVD) were disappointing. 5 Though these early outcome studies often showed reductions in CVD events, no effect on mortality could be demonstrated. This, allied with the adverse effects seen, particularly with clofibrate, led to considerable scepticism about the benefits of lipid-lowering for CVD. The echo of this controversy continues in the popular media to this day. The new statin class of LDL-C lowering therapy with double the efficacy of previous compounds was launched in 1990. 6 The groundbreaking study that resolved the questions about the efficacy of LDL-C lowering in CVD was the Scandinavian Simvastatin Survival Study in 1994. 7 In this study of 4444 patients with chronic coronary heart disease and LDL-C > 5 mmol/l who were treated with simvastatin 20–40 mg to a target LDL-C of 3 mmol/l, statin therapy resulted in a 30% decrease in mortality, and a 30% decrease in CVD mortality, CVD events and interventions as well as hospitalisation. The intervention was also cost-saving to health services. 8 Multiple further trials with statins followed, extending the results to primary prevention [e.g. West of Scotland Coronary Outcomes Prevention Study (WOSCOPS) 9 ], patients with type 2 diabetes (e.g. Collaborative Atorvastatin Diabetes Study; CARDS 10 ) and progressively lower levels of initial LDL-C in each of these sub-groups. All these studies have now been subjected to pooled individual patient meta-analysis, confirming the benefits in all subgroups and establishing a relationship of 21% reduction in CVD events per 1 mmol/l reduction in LDL-C.11,12 All these studies pushed the baseline and post-intervention levels for LDL-C progressively lower, but, as a residual risk of CVD events still existed in treated cohorts, the question were still asked about ‘whether still lower is better’? The only areas where questions remain about the efficacy of LDL-C lowering with statins are in advanced chronic kidney disease (>CKD 4) or established chronic cardiac failure, though sub-groups analysis show benefits in less severely affected population with these conditions. 13
Non-statin therapies and CVD outcomes
Though these studies conclusively identified the benefits of statin therapy, controversy has raged about other interventions. 14 Surrogate outcome studies confused the story with ezetimibe, although eventually an outcomes study in acute coronary syndromes (ACS) with this drug added to statin therapy did finally confirm its utility. In the Improved Reduction of Outcomes: Vytorin Efficacy International Trial (IMPROVE-IT), ezetimibe therapy allowed patients to reach an average LDL-C of 1.4 mmol/l (versus 1.8 mmol/l in statin-alone treated patients) and reduced CVD events by 8% in line with the regression relationship predicted for the degree of LDL-C change from statins. 15 Studies with other drugs such as anacetrapib, which incidentally reduce LDL-C, also followed the same relationship. 16 Currently, the consensus is that any drug intervention that lowers LDL-C is likely to lower CVD events unless it has off-target side-effects. 3
Proprotein convertase subtilisin kexin-9
The search for causes of the genetic defect in FH identified mutations in two genes – the LDL receptor and apolipoprotein B – as causing the majority of cases. However, the search continued for other causes, and mutations in proprotein convertase subtilisin kexin-9 [PCSK9; Neural apoptosis-regulated convertase-1 (NARC-1)] were identified. 17 Further work clarified that these mutations activated the protein, causing functional inactivation (enhanced intracellular degradation) of LDL receptors, whereas other inactivating mutations increasing LDL receptor function were associated with lower LDL-C.18,19 In the Dallas Heart Study, 2.6% of 3363 black patients who had nonsense mutation of PCSK9 leading to a reduction of LDL-C by 28% with better coronary heart disease (CHD) outcomes. 20 A few clinically asymptomatic cases of homozygous PCSK9 deficiency associated with hypolipoproteinaemia have also been described.21,22 These studies laid the theoretical basis for considering intervention to lower LDL-C by targeting PCSK9 (Figure 1).
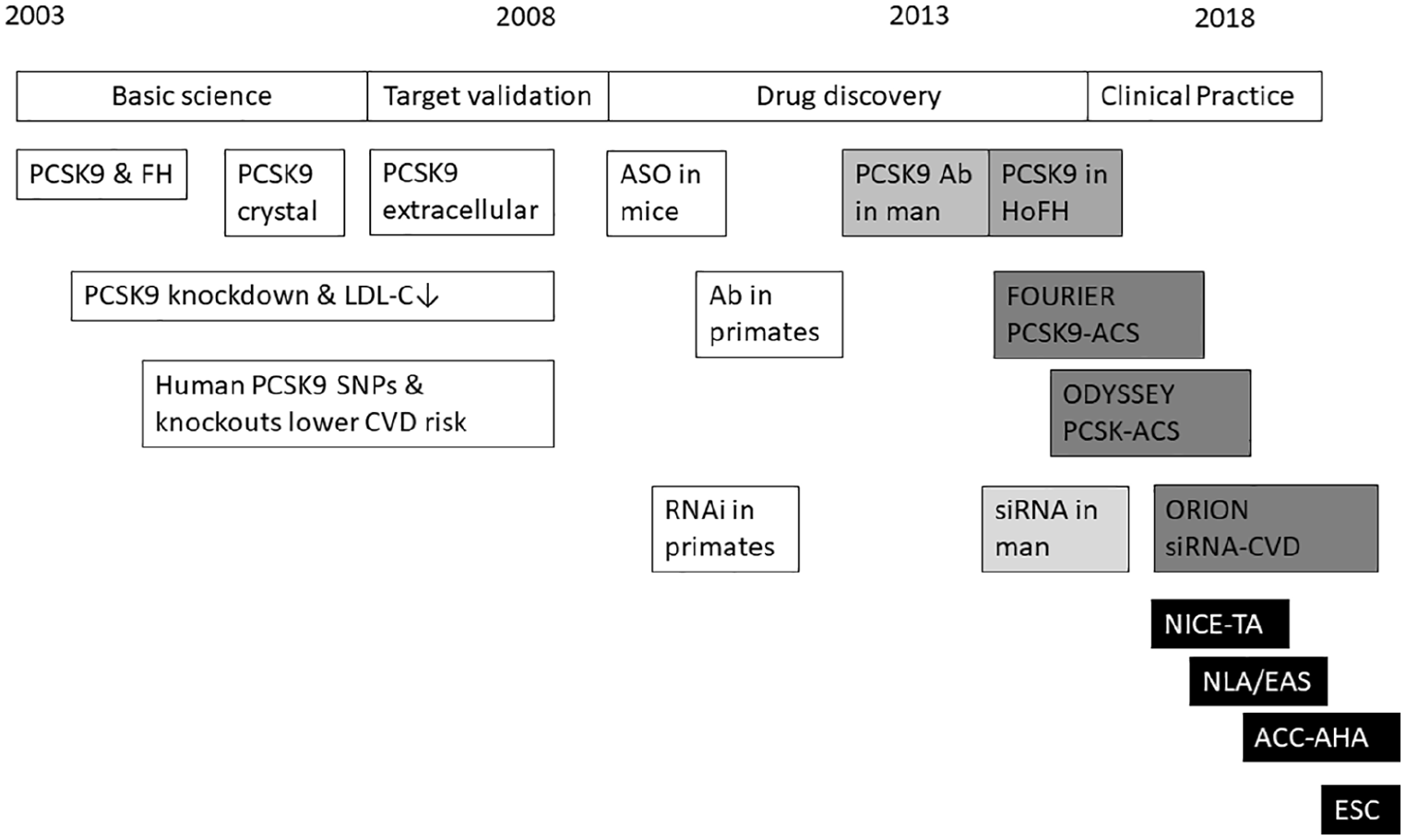
Timeline from PCSK9 discovery to use in clinical practice.
Therapies targeting PCSK-9
Once the role of PCSK9 in controlling plasma LDL-C had been established and there were reasons for suspecting that this intervention would be safe, a systematic search began for compounds that would target this pathway.23–25 PCSK9 exists as a dimer and auto-activates through mutual cleavage of furin-sensitive catalytic domains. The classical approach of small molecule inhibition has proved difficult due to the hydrophobic nature of the compounds required to reach those binding sites, 26 whereas other approaches remain exploratory. 27 Many of these hydrophobic molecules have poor bioavailability as oral compounds need to be water-soluble and for food (fat) effects to be limited to allow licensing. 27 Though no cases of autoimmune-based hyper- or hypolipoproteinaemia due to anti-PCSK9 antibodies have been described, animal studies showed that human PCSK9 was antigenic and this allowed the development of a series of antibody-based therapies based on humanised (-zumab) or human (-cumab) antibodies.
Alirocumab and Evolocumab are fully human anti-PCSK9 antibodies and are licensed for clinical practice as they reduce LDL-C by 54% when given fortnightly. 28 As with all antibody therapies, their adverse effects tend to be related to the structure of the antibody, hence causing increases in injection site reaction [1.51 versus 0.83 per 100 patient-years; relative risk (RR) 1.41, 95% confidence interval (CI) 1.21–1.65); p < 0.001], but also in practice symptoms of flu-like myalgia post-injection (up to 10%).25,28,29 No compound-specific adverse events have yet been described. High expression of PCSK9 in the cerebellum and gut exists, 30 and it has been suggested that these drugs may be associated with neurological adverse effects, especially neurocognitive deterioration, though again nothing has been seen in the clinical trials to date [0.57 versus 0.55 per 100 patient-years; RR 1.01 (0.84–1.21); p = 0.91].28,31
PCSK9 inhibitors and new-onset diabetes
It took 15 years for the effect of statins in increasing blood glucose by approximately 0.3 mmol/l and hence rates of new diabetes to be noticed. 32 Mendelian randomisation studies that validated the association of variants in the 2-hydroxy-methyl-glutaryl-CoA (HMG-CoA) reductase gene with increased rates of diabetes, possibly related to an increase in body weight, 33 have also suggested that PCSK9 inhibition may cause similar problems. 34 A meta-analysis of 68,123 patients from 20 trials with median follow up of 78 weeks suggested that PCSK9 inhibitors increased fasting blood glucose (1.88 mg/dl; 0.1 mmol/l; p = 0.001) and HbA1c by 0.03% (p < 0.001) when compared with placebo but, over a 2-year timescale, this was not sufficient to increase incidence of diabetes [RR 1.04 (0.96–1.13); p = 0.43]. An exploratory meta-regression analysis suggested an increased risk of diabetes with the potency (p = 0.03) and duration (p = 0.03) of PCSK9 inhibitor therapy. 35 However, in a meta-analysis of 39 trials with a weighted follow-up time of 2.3 years comprising 150,617 patient-years, the rate of new-onset of diabetes mellitus was unchanged [1.92 versus 1.93 per 100 patient-years (RR 1.00 (0.93–1.07); p = 0.97]. 28 Similar results were seen in the long-term clinical CVD outcome studies of these drugs.28,35 In a pre-specified analysis of the ODSSEY trial by glycaemic status, alirocumab resulted in similar relative reductions in the incidence of the primary endpoint in each glycaemic category. Among patients without diabetes at baseline, no difference was seen in new onset diabetes [10.1 versus 9.6%; (HR 1.00 (0.89–1.11]. 36 In the FOURIER study, 11,031 patients (40%) had diabetes, 10,344 had pre-diabetes and 6189 were normoglycaemic. No increase was seen in new-onset diabetes in patients without diabetes at baseline (HR 1.05 (0.94–1.17), including in patients with pre-diabetes (HR 1.00 (0.89–1.13). Levels of glucose and HbA1c were similar between evolocumab and placebo over time in all groups. 37
Antibodies to PCSK-9
However, the humanised antibody, Bococizumab, which contains murine sequence, although initially delivering a 55% reduction in LDL-C like other antibodies, has not been licensed following longer-term studies. This was caused by the attenuation of its effect secondary to development of anti-drug antibodies in the outcome trials (SPIRE-1 and SPIRE-2) with a frequency of 48% after 3 months resulting in titre-dependent attenuation of drug effect.38,39 Though anti-drug antibodies have been described for human PCSK9 antibodies, none have been associated with loss of efficacy, yet there are growing anecdotal reports of patients who do not respond to PCSK9 therapy. 40 Whereas many of these non-responders have disturbances in lipid metabolism associated with increased synthesis of very-low-density lipoprotein (VLDL), and hence LDL-C, and thus logically might not respond to a drug that increases recycling of cholesterol back to the liver through the LDL receptor and so require additional treatment to correct this problem, 40 some seem to be primary non-responders. No relationship with anti-drug antibodies has been shown in the few cases investigated though potentially enhanced cell-mediated reticulo-endothelial clearance of PCSK9 antibodies through Fc receptors has not been investigated.
PCSK-9 development trials
All PCSK-9 inhibitors have followed the standard phase I–III development programme used for lipid-lowering therapies. 41 After individual dose, multiple dose and dose ranging studies, phase III trials have been conducted in patients with/without CHD, familial hypercholesterolaemia (FH), and type 2 diabetes either compared with placebo, added to baseline statins or in comparison with ezetimibe. 28 Data are still awaited on populations with chronic renal failure and human immunodeficiency virus (HIV). The most interesting studies were in homozygous FH (HoFH) and statin intolerance. Statins, in contrast to ezetimibe, show significantly reduced efficacy in HoFH and do not work in patients with null mutations. Initial studies with PCSK9 inhibition showed substantially preserved efficacy in patents with HoFH due to point mutations in the LDL receptor, reduced efficacy in patients with a single null allele and no response in dual null allele patients. 42 These findings were replicated in larger studies.43,44 Up to 25% of patients with HoFH can discontinue apheresis, 45 though in most cases apheresis has been continued and LDL-C targets have been adjusted to those used in routine practice.
Statin intolerance is a substantial problem, with a claimed prevalence of 10% (although in reality 1% in re-challenge studies). 46 It is defined as inability to continue to take a statin despite trying three drugs at different doses.47,48 Two studies have been conducted with PCSK9 inhibitors in this population. In the GAUSS-2 trial, 307 statin-intolerant patients with LDL-C 4.97 mmol/l were randomised to evolucumab or ezetimibe. 49 Evolocumab reduced LDL-C by 53–56%, compared with 14–16% for ezetimibe. Muscle adverse events occurred in 12% of evolocumab-treated patients and 23% of ezetimibe-treated patients. The larger GAUSS-3 study in 511 patients reproduced these effects. 50 In the ODYSSEY-alternative study, 361 statin-intolerant patients received an injection and oral placebo for 4 weeks during placebo run-in. 51 Patients (13%) reporting muscle-related symptoms during the run-in were to be withdrawn. Continuing patients were randomised (2:2:1) to alirocumab 75 mg fortnightly (with option to increase), ezetimibe 10 mg, or atorvastatin 20 mg/day with appropriate placebos for 24 weeks and then open follow up. The baseline LDL-C was 5.0 mmol/l. Alirocumab reduced LDL-C by 52% versus 17% with ezetimibe. Treatment was discontinued by 18% with alirocumab and 25% for ezetimibe or statin therapy. Skeletal muscle-related events were seen in 16% with alirocumab compared with 20% with ezetimibe and 22% with atorvastatin. Open label statin was well tolerated by those patients who were unblinded at study completion.
PCSK-9 cardiovascular outcomes trials
There are increasing challenges in designing outcome trials in CVD. Progressive lowering of acceptable LDL-C levels for recruitment into studies as a result of guidelines and ethical considerations leads to lower event rates and thus potentially larger trial populations. It is now increasingly common to concentrate on high-risk sub-groups such as patients with ACS and to require the presence of additional CVD risk factors such as increased age, smoking, or type 2 diabetes. All of these changes make the trials possible in terms of timescales, logistics, and finance, but cause difficulties for guideline groups that have to generalise the findings outside the original trial population. The recent PCSK9 trials follow these design precepts.
FOURIER trial: evolocumab and CVD outcomes
The FOURIER recruited 27,564 patients with a history of recent atherosclerotic vascular disease (cerebrovascular, cardiovascular and peripheral artery disease) but also an additional one major or two minor risk factors allied with a LDL-C > 1.8 mmol/l (70 mg/dl) or nonHDL-C > 2.6 mmol/l (100 mg/dl). 52 Additional risk factors included hypertension (80%), type 2 diabetes (37%) and smoking (29%). These patients had to be receiving statin therapy of at least 20 mg of atorvastatin with or without ezetimibe, but only 70% were on high-intensity statin. In reality, given questions about the efficacy of ezetimibe at the time, 5% were on this treatment. Patients were randomised to evolocumab injections (either 140 mg every 2 weeks or 420 mg every 4 weeks) or matching placebo. The primary endpoint was a combination of fatal and non-fatal myocardial infarction (MI) and stroke (cerebrovascular accident; CVA) including unstable angina (UAS)warranting hospital admission or coronary revascularisation. The secondary outcome excluded revascularisation episodes. LDL-C decreased by 59% from 2.2 mmol/l (92 mg/dl) to 0.8 mmol/l (30 mg/dl) in the evolocumab group, while non-HDL-C levels decreased by 52% and ApoB levels by 49%. Evolocumab reduced the five-point primary endpoint (9.8% versus 11.3%; HR 0.85; p < 0.001) and secondary endpoint (a composite of CVD death, MI, or CVA) (5.9% versus 7.4%; HR 0.80; p < 0.001) at a median follow up of 2.2 years. This was driven by reduction in the risk of non-fatal MI, CVA and coronary revascularisation. Few serious adverse events were found, with no difference in new-onset diabetes or neurocognitive events. However, injection-site reactions were more common with evolocumab (2.1% versus 1.6%).
Major CVD events declined progressively with decreasing LDL-C concentrations achieved, with adjusted HR in the group with LDL-C < 0.2 mmol/l (10 mg/dl) of 0.69 for the primary and 0.59 for secondary endpoints compared with those with LDL-C > 2.6 mmol/l (100 mg/dl). 53 High-risk subgroups such as those with differing extent of coronary artery disease, 54 type 2 diabetes mellitus 37 or peripheral arterial disease 55 had higher event rates and consequently greater absolute benefits with evolucumab therapy.
ODYSSEY-outcomes: alirocumab and CVD outcomes
The ODYSSEY Outcomes trial randomised 18,924 patients with history of an ACS within the previous 12 months to either alirocumab 75 mg or placebo every 2 weeks. Inclusion criteria included either LDL-C > 1.8 mmol/l (70 mg/dl), nonHDL-C > 2.6 mmol/l (100 mg/dl) or ApoB > 80 mg/dl after maximal statin therapy (atorvastatin 40–80 mg or rosuvastatin 20–40 mg). 56 The primary composite end-point included death from CAD or CVD, myocardial infarction, ischemic stroke or angina requiring hospital admission. Most patients (93%) were enrolled due to LDL-C > 1.8 mmol/l (70 mg/dl). The on-treatment LDL-C target was 0.6–1.3 mmol/l (25–50 mg/dl). Alirocumab was up-titrated from 75 mg to 150 mg every 2 weeks in patients who had LDL > 1.3 mmol/l (50 mg/dl). Conversely, patients who had LDL-C levels consistently below 0.2 mmol/l (15 mg/dl) were switched to placebo. After a median follow up of 2.8 years, LDL-C levels had decreased by 55% to 1.3 mmol (53 mg/dl) from 2.4 mmol/l (101 mg/dl). The primary endpoint was significantly lower in the alirocumab group compared with the placebo group (9.5% versus 11.1%, HR 0.85, p = 0.0003) after a median 2.8 years, with benefit seen only after 1 year of treatment as in statin trials. The reduction in MACE with alirocumab was primarily because of reduction in ischaemic events: nonfatal MI was reduced by 14%, CVA by 27% and UAS by 39%. All-cause mortality was reduced by 15% (3.5% versus 4%, p = 0.03) but CAD (2.2% versus 2.3%) and CVD (2.5% versus 2.9%) deaths were similar. Minor injection site reactions were commoner with alirocumab (3.1 versus 2.1%). In patients with baseline LDL-C > 2.6 mmol/l (100 mg/dl) comprising 30% of total, CVD events were reduced by 24%, with all endpoints showing reductions, including CAD death and CVD death by 28% and 31%, respectively.
In a pre-specified analysis of ODYSSEY Outcomes study, the total number of nonfatal CVD events and deaths prevented with alirocumab was twice the number of first events prevented. 57 In this study, 3064 first and 5425 total events occurred in the whole cohort. In the alirocumab arm, there were 190 fewer first and 385 fewer total nonfatal CVD events or deaths. Alirocumab thus decreased the occurrence of first, but also subsequent, CVD events.
An overview of PCSK9 outcome trials
Both trials reduced primary composite CVD end points. The Kaplan–Meier curves diverged after 12 months and the effects accumulated as the length of treatment extended. The incidence of primary composite end points in the FOURIER trial decreased by 15 (12–27)%, similar to the 12 (7–16)% achieved during the first year of treatment with statins in the Cholesterol Treatment Trialists’ (CTT) meta-analysis. 52 Similar results were obtained in ODYSSEY outcomes, indicating that the CVD event, i.e. change in LDL-C relationship (21% relative risk reduction per 1 mmol/LDL-C reduction), held for these drugs as well as statins and ezetimibe.
As usual for lipid trials, the greatest effects with PCSK9 inhibitors were seen on coronary intervention and non-fatal outcomes. 58 In a meta-analysis of 83,321 patients, PCSK9 inhibitor therapy did not reduce all-cause mortality [RR 0.94 (0.81–1.09); p = 0.41], even after excluding the bococizumab studies. 59 Predictably, this risk reduction varied with baseline LDL-C, in showing a significant reduction only in patients with LDL-C > 100 mg/dl (2.4 mmol/l) [RR 0.39 (0.20–0.76); p = 0.01]. PCSK9 inhibitor therapy showed no significant effect on CVD mortality, but, after regrouping ODYSSEY OUTCOME estimates, again an effect restricted to patients with LDL-C > 100 mg/dl (2.4 mmol/l) [RR 0.67 (0.51–0.87); p = 0.006] was detected. 59
Concerns have been expressed about the association of low LDL-C with excess CVD risk, especially for haemorrhagic stroke. 60 The PCSK-9 endpoint trials allowed this to be explored, though the ODYSSEY trials did have a compulsory dose reduction if achieved LDL-C was 0.5 mmol/l (20 mg/dl). In the FOURIER study, 8003 (31%) patients achieved concentrations of 0.5–1.3 mmol/l. 53 The relationship between lower LDL-C and lower risk of CVD endpoints continued and extended to the bottom first percentile (LDL-C < 0.2 mmol/l. No excess adverse events, including haemorrhagic strokes, were seen. This confirms the safety of achieving ultra-low LDL-C in patients with ACS.
Insights of PCSK-9 trials into optimal targets
Imaging-based studies have a long history in CVD studies. Originally studies were preformed using quantitative coronary angiography, and these showed the benefits of statin, fibrate, niacin and bile acid sequestrant treatment as well as strict LDL-C-lowering diets. The technology evolved and later trials were conducted using intravascular ultrasound (IVUS) in patients with one 50% coronary artery stenosis over a 2-year time period. The first trial using this methodology was the Reversal of Atherosclerosis With Lipitor (REVERSAL) trial comparing Pravastatin 40 mg with Atorvastatin 80 mg. 61 This data was later analysed by achieved LDL-C, and regression began to be seen with LDL-C < 1.8 mmol/l.62,63
Other groups performed similar studies with ezetimibe added to statin therapy. The largest is the plaque regression with cholesterol absorption inhibitor or synthesis inhibitor evaluated by intravascular ultrasound (PRECISE-IVUS) trial in 202 patients with previous CAD intervention. 64 Statin therapy was optimised to deliver LDL-C < 1.8 mol/l and patients were randomised to ezetimibe or placebo for 1 year and followed by IVUS imaging. The achieved LDL-C was 1.6 mmol/l (63 mg/dl) compared with 1.9 mmol/l (73 mg/dl) resulting in percent atheroma volume (PAV) change of 1.54 (0.003–3.08)%, which did not exceed the pre-defined noninferiority margin of 3%. The absolute change with statin-ezetimibe in PAV was −1.4% versus –0.3% with statin alone (p = 0.001) and 78% versus 58% (p = 0.004) patients showed evidence of regression.
A similar study has been conducted with a PCSK-9 inhibitor. The global assessment of plaque regression with a PCSK9 antibody as measured by intravascular ultrasound (GLAGOV) trial randomised 968 patients aged 60 years with angiographic CAD to 420 mg monthly or placebo on top of statin therapy for 76 weeks. 65 The evolucumab group achieved a LDL-C of 0.9 mmol/l (37 mg/dl) compared with 2.2 mmol/l (93 mg/dl) in the placebo group. Evolocumab treatment reduced PAV by 1.0% overall, inducing 0.95% regression compared with a 0.05% increase in the placebo group. In 144 patients with LDL-C levels < 1.8 mmol/l (70 mg/dl) the change in PAV was more marked (−1.97% versus −0.35%; p < 0.001), with 81% of patients on evolocumab showing plaque regression. The study confirmed that evolocumab added to statin therapy induced significant plaque regression.
Health economics of PCSK-9 therapy
The main problem with PCSK-9 therapies has been their substantial price differential compared with other agents used in CVD medicine. 66 Though priced similarly to other antibody-based therapies, this has provoked resistance from payers and caused controversy within guidelines groups. Health economic analyses show that off-patient drug therapies are highly cost-effective in primary prevention, whereas in secondary prevention on-patent therapies costing about $100/month are cost effective in ACS but not chronic CVD if a low threshold of $30,000 ($30k) per quality adjusted life year (QALY) is used as in the United Kingdom (UK). 67 Using thresholds more typical of the United States (US) at $50–100k/QALY, these drugs may be cost effective in higher-risk secondary prevention but are limited in primary prevention.68,69 Analyses using the initial process of PCSK-9 inhibitors suggested poor cost-effectiveness at a $150k threshold unless optimistic assumptions were made.70,71 Value-based pricing analyses in the US suggested that a price in the region of $5–6k was reasonable given their benefits. 70 Prices for PCSK-9 inhibitors have since been reduced in the US from $15–16k per year to a new global price in the region of $6k, rendering these drugs more acceptable to payers. European payers have negotiated further discounts to improve the acceptability of these drugs in social insurance/government-funded health systems, but concerns about price still limit access to these medications in many countries.69,72
The outcome of these concerns about the affordability of PCSK-9 inhibitors has been a tendency towards underuse of these drugs compared with specialist society guideline recommendations at 1–2% of the population concentrated on the highest risk groups.73–75 However, significant differences are likely to arise between payer-derived guidelines [e.g. UK National Institute for Health and Clinical Excellence (NICE)] and specialist societies that do not need to consider reimbursement. 76 The gap between clinical desires and financial affordability opens the way to alternative methods of lowering LDL-C.
Alternative approaches to lowering LDL-C
Data from imaging and CVD outcomes studies suggest that most therapies that reduce LDL-C will also reduce CVD events. This has encouraged the development of further compounds that reduce LDL-C.
Reduction of LDL-C with small interfering RNA therapy: inclisiran
An alternative approach to inhibiting PCSK9 in plasma with antibodies is to inhibit translation of the protein in the cytoplasm by interfering with the messenger RNA (mRNA). 77 Theoretically, this has the advantage that this will inhibit the intra-cellular activities of PCSK9, whereas antibodies only affect the extracellular compartment. Two approaches exist to inhibit the mRNA. The longer established strategy in lipids is to inhibit mRNA translation through the use of subcutaneously administered antisense oligonucleotides (ASOs). The alternative is to use short interfering RNA (siRNA). The siRNA blocks translation of PCSK9 mRNA and promotes RNA degradation through RNA-induced silencing complex (RISC), in contrast with ASOs, which use a RNAse H1-based degradation pathway.77,78 Further targeting specificity can be added through a n-acetyl-glucosamine (NAG) moiety that targets the drug to hepatocytes, enabling siRNA doses to be reduced compared with untargeted ASOs. Unfortunately, ASO approaches to PCSK9 have proved unsuccessful due to high rates of injection-site reactions and renal tubular toxicity. 79
Inclisiran is a long-acting siRNA molecule targeting PCSK9 mRNA. A dose-ranging study of several doses of inclisiran in adult healthy volunteers with an LDL-C > 2.6 mmol/l (100 mg/dl) showed that multiple-dose regimens of inclisiran reduced levels of PCSK9 and LDL-C by 84% and 60% from baseline at day 84. 80 Doses ⩾300 mg reduced PCSK9 and LDL-C levels for at least 6 months. No serious adverse events were seen with inclisiran, and the most common adverse events in the trial were cough, musculoskeletal pain, nasopharyngitis, headache, back pain and diarrhea. This study prompted further development of inclisiran. In the ORION-1 study of 501 patients, inclisiran produced dose-dependent reductions in PCSK9 levels and LDL-C. 81 The greatest reduction in LDL-C was seen with two-doses of 300 mg inclisiran regimen (first dose at day 1 and second dose at day 90), resulting in 48% of patients achieving LDL-C < 1.2 mmol/l (50 mg/dl) at 6 months to 1 year. 82 Various phase III studies with inclisiran are ongoing. The ORION-4 study is randomising 15,000 patients aged >55 years with pre-existing CVD to inclisiran or placebo. 83 They will be followed up for 5 years for safety and efficacy in preventing major adverse CVD events. More than 1550 patient-years of safety data have been accumulated in the ORION phase III program with no significant adverse safety signals.
PCSK9 vaccines
As PCSK9 is antigenic and can be used to produce human antibodies, then it is feasible to attempt a long-term vaccine strategy to accomplish similar effects on a permanent basis. Initial experimental results in animals vaccinated with bacteriophage virus-like particle (VLP) conjugated with PCSK9 epitopes adjacent to the LDLR binding domain have been encouraging. 84 Immunisation against PCSK9 in various mouse models has been shown to decrease cholesterol and LDL-C levels, reduce development of CVD, and decrease systemic and vascular inflammation. 85 However, no human studies have yet been performed.
PCSK9 gene therapy
Individuals homozygous for PCSK9 deficiency have been identified and have clinical phenotypes of being healthy though profoundly hypocholesterolaemic. 21 Thus, clinical intervention to knock out PCSK9 using gene therapy technology such as Clustered regularly interspaced short palindromic repeat-(CRISPR)-associated protein -9 (CRISPR-Cas9) may be feasible to deliver a cure for conditions such as FH.86,87 Again, no studies in man have yet been performed.
Bempedoic acid
As there is considerable confidence in the LDL-C hypothesis, and as statins target the cholesterol synthesis pathway, it may be possible to further inhibit this pathway. 88 Originally, studies concentrated on squalene synthase inhibition later in the pathway, but these drugs (e.g. zaragozic acid, lapaquistat) proved to have limited efficacy and potentially high toxicity. 89
The newest development is to target the step before HMG-coA reductase in the liver: ATP-citrate lyase. Bempedoic acid is an oral inhibitor of hepatic ATP citrate lyase, which decreases LDL-C levels by 17% in patients receiving maximally tolerated statin therapy at 12 weeks without obvious serious adverse events. 90 In statin-intolerant patients, bempedoic acid added to other lipid lowering therapy, including ezetimibe, was well tolerated and resulted in 24% lower LDL-C compared with placebo or when added to ezetimibe therapy again after 12 weeks. 91 Longer-term safety has been demonstrated in 2230 patients. 92 This drug is now being evaluated in a CVD outcomes study.
Implications of PCSK-9 trials for guidelines
Lipid guidelines have generally followed the 50th centile of achieved LDL-C from the CVD outcome trials as a target. Thus, targets fell rapidly from 3 mmol/l in secondary prevention (4S) to 1.8–2.0 mmol/l (CARE and LIPID studies). 76 The high-dose compared with low-dose statin studies in secondary prevention reinforced the 1.8–2.0 mmol/l target especially after the CTT meta-analysis was published, but did not reduce the targets further despite the analysis showing continuing linear relative risk reduction down to 1.5 mmol/l. The results of recent studies, such as IMPROVE-IT or the PCSK-9 trials, in ACS are just beginning to be implemented in the guidelines.93,94 Almost all new guidelines have agreed that ezetimibe should be added to statin therapy in ACS as it is moderately effective and off-patent, and thus cheap.93,94 UK NICE is the only exception as its appraisal is dated.
Incorporation of PCSK-9 trials into guidelines has proved problematic, due primarily to cost considerations. Specialist societies later followed by guidelines have based their recommendations on the trial populations by creating the concept of high-risk groups of patients with CVD distinguished by excess vascular risk (ACS or multi-vascular bed disease) hyperlipidaemia and additional risk factors.95–97 The American and European society guidelines,93,94 like their specialist lipid societies, introduced the concept of ultra-high-risk groups based on the secondary prevention populations recruited to trials, whereas others have suggested the use of number needed to treat (NNT) thresholds, which are a proxy for CVD risk. 98 Patients with FH or recurrent ACS, LDL-C > 2.5 mmol/l and additional risk factors qualify for treatment. European guidelines have gone further by extending the concept to ultra-high primary prevention. UK NICE is conservative in preserving the concept of one category of established CVD and suggesting prescription in FH with LDL-C > 5 mmol/l or patients with CVD and LDL-C > 4 mmol/l for monovascular or >3.5 mmol/l in multi-vascular bed or recurrent disease in patients despite maximally tolerated statin and ezetimibe.
These guidelines might be thought to substantially increase the population eligible for PCSK-9 therapy. Currently, the UK NICE guidelines suggest 2% of patients with CVD may be eligible for PCSK9 inhibitors despite 29% not achieving LDL-C targets of <2 mmol/l, 99 whereas a Canadian analysis suggests up to 52% may require PCSK-9 therapy. 72 However, in both FOURIER and ODYSSEY, lipid-lowering therapy was not maximised prior to randomisation and ezetimibe usage was low. In IMPROVE-IT, baseline lipid levels were similar to the PCSK-9 trials at a LDL-C of 2.43 mmol/l (94/dl), but optimisation of statin therapy (simvastatin only) reduced this to 1.81 mmol/l (67 mg/dl), whereas addition of ezetimibe allowed a mean LDL of 1.38 mmol/l (53 mg/dl) to be achieved, in line with suggested targets from both major guidelines. The only major difference between these guidelines is in the new European guidelines, which suggests a lower target of 1 mmol/l for LDL-C in patients with recurrent ACS within 1 year of initial intervention based on the PCSK9 trials.
All the guidelines and some advisory strategies concentrate on lipid-lowering as part of their brief, in effect focusing on NNT and, hence, underlying CVD risk. 98 These approaches neglect the substantial benefits seen on CVD mortality and events seen in some sub-groups from non-lipid-lowering-based therapies. Sodium-glucose lithium transporter-2 (SGLT-2) inhibitors reduce mortality, heart failure and CVD events in patients with diabetes, 100 whereas glucagon-like peptide-1 (GLP-1) agonists reduce CVD events. 101 These patients may encompass 25–30% of eligible patients based on PCSK-9 trial recruitment. Furthermore, other interventions such as high-dose omega-3 fatty acids (reduction of cardiovascular events with EPA-intervention trial; REDUCE-IT) in patients with moderate hypertriglyceridaemia (which overlap with PCSK-9 trial recruitment criteria) also reduce CVD events. 102 Other data suggest that more aggressive anti-thrombotic therapy or even colchicine therapy may further reduce the eligible population by reducing CVD events from other causes without needing to prescribe expensive drugs. 103 Thus, the eligible population for PCSK9 inhibitors is, in reality, patients with hypercholesterolaemia who are statin-intolerant or those with FH.
Conclusion
PCSK-9 inhibitors are a major innovation in lipid management and are undoubtedly effective in reducing both CVD events and LDL-C by 50% with acceptable rates of discontinuation. Uptake of these drugs has been slow, driven by concerns about affordability from payers, but increasing flexibility on pricing is helping to resolve this in many countries. New PCSK-9 interventions are in development, as are alternative oral methods of lowering LDL-C. Though PCSK-9 inhibitors will undoubtedly have a place in lipid management, it is unclear as to whether they will be universally used or just prescribed to selected high-risk groups.