
Abstract
There is a global obesity epidemic that will continue to be a financial burden on healthcare systems around the world. Tackling obesity through diet and exercise should always be the first intervention, but this has not proved to be effective for a large number of patients. Pharmacotherapeutic options have been limited and many previously available drugs have been withdrawn due to safety concerns. Currently, only bariatric surgery has the capability to induce both substantial and durable weight loss. This article briefly reviews the history of pharmacotherapy for obesity before focusing on the clinical trial evidence for the use of the GLP-1 agonist liraglutide as a weight loss agent and comparing its efficacy with other emerging drug therapies for obesity.
Introduction
The trend towards reduced physical activity and increased calorific value of diets in the modern world has led to a pandemic of overweight and obesity. The World Health Organization (WHO) estimates a worldwide prevalence of overweight [a body mass index (BMI) of 25 kg/m2 or higher] of 38% of men and 40% of women [World Health Organization, 2015]. Overweight and obesity are associated with many negative health outcomes including type 2 diabetes mellitus [Colditz et al. 1995], hypertension [Must et al. 1999], cancers [Eheman et al. 2012], cardiovascular disease [Asia-Pacific Cohort Studies Collaboration, 2006], osteoarthritis [Davis et al. 1990], depression [Simon et al. 2008] and other comorbidities [Guh et al. 2009]. Importantly, obesity also carries a significant quality of life cost [Soltoft et al. 2009]. The mortality hazard for a person with BMI of 40 kg/m2 is approximately twice that for someone of normal BMI (BMI 18.5 to 25kg/m2), even after adjustment for the comorbidities associated with being overweight [Berrington et al. 2010]. In one prospective cohort study, an obese 40-year-old woman nonsmoker lost 7.1 years of life expectancy compared with their normal weight counterpart, with a loss of 5.8 years in men [Peeters et al. 2003]. Furthermore, the financial cost of obesity, both in terms of direct healthcare liabilities and indirect economic impact, is enormous and projected to be as much as £27 billion lost to the UK economy in 2015 [Morgan and Dent, 2010].
It is well established that weight loss in obese patients results in both improvement in comorbidities [Wing et al. 2011] and reduction in all-cause mortality [Kritchevsky et al. 2015]. The challenge is achieving this target in a durable way. Lifestyle modifications involving low-calorie diets and/or exercise are effective in inducing weight loss over the short term but the tendency is for much of the lost weight to be regained over time [Franz et al. 2007]. Thus, there is a need for effective additional interventions to provide long-term weight control. Bariatric surgical procedures have been shown to be efficacious [Sjostrom et al. 2012] and cost effective [Picot et al. 2009]. Perioperative morbidity and mortality is low [Aminian et al. 2015], but it is not always possible to avoid the sometimes persistent side effects of post-prandial nausea or vomiting, micronutrient deficiencies or hypoglycaemia related to dumping syndrome. Uptake of surgery in those eligible for this intervention is likely to be limited: the 2007 National Institute for Health and Clinical Excellence (NICE) guide for the commissioning of bariatric surgery assumed a take-up rate of 30–50% [National Institute for Clinical Excellence, 2007]; Since then, NICE has updated the guidelines for the management of obesity and in the absence of other effective measures, has widened the criteria for those eligible [National Institute for Health and Clinical Excellence, 2014a]. There are now estimated to be approximately 1 million people in the UK potentially eligible for surgery [National Institute for Health and Clinical Excellence, 2014b]. It is likely that relatively few of those in this new patient group will opt for surgery. Finding an effective, nonsurgical means of achieving an equivalent weight loss, and its associated health gain, for these patients is thus an urgent priority.
History of pharmacotherapy for obesity
The history of pharmacotherapy for obesity is a chequered one. The first licensed drugs, amphetamine derivatives, were approved by the nascent US Food and Drug Administration (FDA) in the 1940s. They remain available for use in the US but the addictive properties of amphetamines allow for short-term use only, limiting their efficacy for long-term weight loss. By the 1970s their use had dwindled. In an effort to reduce the side-effect burden but retain weight-loss efficacy, combination tablets of lower doses of amphetamine derivatives were explored. Phentermine–fenfluramine (or ‘phen-fen’) was marketed in the early 1990s after a successful trial of 121 patients showing a placebo-subtracted weight loss of 11.0% of baseline without frequent or serious adverse effects [Weintraub et al. 1992]. It became very popular and was heavily used, but had to be withdrawn from the market in 1997 due to evidence linking fenfluramine first with pulmonary hypertension [Brenot et al. 1993] and then with valvulopathies [Connolly et al. 1997]. The same year, sibutramine, a dual serotonin–noradrenaline reuptake inhibitor with anorectic and thermogenic effects [Luque and Rey, 2002], was licensed for use in the US and Europe. However, in 2010 evidence of an increased risk of myocardial infarction and stroke was published [James et al. 2010] and led to the European Medicines Agency (EMA) withdrawing approval.
Rimonabant, an inverse agonist of the cannabinoid receptor CB1, was approved by the EMA (although not the FDA) in 2006. The phase III RIO trial had shown a signal toward increased psychiatric adverse events despite those with psychiatric disorders being excluded from the trial [van Gaal et al. 2005]. A detailed review of the incidence of suicidality in clinical trials of rimonabant, undertaken by Sanofi-Aventis at the request of the FDA, found an odds ratio of 1.9 [95% confidence interval (CI) 1.1–3.1] on a dose of 20 mg compared with placebo [US Food and Drug Administration Advisory Committee, 2007] and it was withdrawn from the European market. Between 2010 and 2015 physicians in the EU had only one licensed drug available for use: orlistat, a pancreatic lipase inhibitor which reduces absorption of dietary lipids. Orlistat results in an approximate 3 kg excess weight loss over placebo per year [Padwal et al. 2003] and remains widely prescribed. The XENDOS trial found that the addition of orlistat to lifestyle measures reduced progression to type 2 diabetes in obese patients with impaired glucose tolerance by 45% over 4 years [Torgerson et al. 2004]. Unfortunately, its mechanism of action leads to gastrointestinal side effects which sometimes prove intolerable and which have somewhat limited its use.
New anti-obesity pharmaceuticals
On the background of this chequered history, several new drugs have recently emerged. A serotonin mimetic called lorcaserin (Belviq®), which possesses appetite suppressant properties through its action at 5-HT2c receptors on pro-opiomelanocortin (POMC) neurons in the appetite centres of the hypothalamus [Thomsen et al. 2008], received FDA approval in 2012. However, the application for authorisation from the EMA was withdrawn after concerns were raised regarding potential negative effects on psychiatric and cardiovascular risk. A phentermine/topiramate combination tablet (Qsymia®) was initially refused authorization by both the FDA and EMA on the grounds of uncertainties as to the cardiovascular safety of phentermine and psychiatric/neurocognitive effects of topiramate [European Medicines Agency, 2013], but after further safety data were provided it was approved in the US in 2012. The mechanism for the weight loss effect of topiramate is uncertain but is likely to involve both reduced calorific intake and increased energy expenditure [Allison et al. 2012].
Within the last year, two further medications for obesity have been approved. Bupropion/naltrexone (Contrave® in US, Mysimba® in EU), was authorized by the FDA in September 2014. Bupropion stimulates the same POMC neurons as lorcaserin through incompletely understood mechanisms and naltrexone blocks autocrine downregulation of this activity by antagonizing opioid receptors [Greenway et al. 2009]. Soon after, in December 2014, the FDA approved liraglutide (under the new name Saxenda®), a glucagon-like peptide-1 (GLP-1) receptor agonist, familiar as a treatment for type 2 diabetes mellitus, for use as a weight loss agent in patients without diabetes. EMA approval for bupropion/naltrexone and liraglutide arrived in March 2015 and the drugs now await member state authorization.
This article will focus on liraglutide, its effects on weight, and how physicians might use high-dose liraglutide in the treatment pathway for patients with obesity.
Previous use of GLP-1 agonists
An oral glucose load results in a greater and more sustained insulin secretion than does an equivalent intravenous load [Elrick et al. 1964], a phenomenon known as the incretin effect. Endogenous GLP-1 was discovered in the early 1980s [Bell et al. 1983] and was shown to act as an incretin at physiological concentrations [Kreymann et al. 1987]. In vivo, GLP-1 is rapidly broken down by dipeptidyl peptidase-4 (DPP-4) and has a brief half-life of 2 minutes, making it unsuitable as a pharmacotherapy for diabetes. However, the potential for pharmacological manipulation of the GLP-1 receptor was evident. The emergence of longer-acting receptor agonists began with the discovery of exendin-4 from the venom of the gila monster, Heloderma suspectum [Eng et al. 1992]. Exendin-4 is a peptide with 53% amino acid identity to human GLP-1. It was shown potently to stimulate the GLP-1 receptor and induce proinsulin gene expression in islet beta cells [Goke et al. 1993]. It is resistant to DPP-4 degradation and possesses a much longer half-life than GLP-1 [Parkes et al. 2001], resulting in prolonged glucose-lowering activity in vivo [Young et al. 1999]. It was soon recognized that, in contrast to previously available insulin secretagogues such as sulfonylureas, administration of exendin-4 resulted in weight loss in animal models [Young et al. 1999], an effect that was later reproduced in phase III clinical trials [DeFronzo et al. 2005]. Exenatide, a synthetic peptide identical to exendin-4, was the first GLP-1 receptor agonist approved for the treatment of type 2 diabetes, authorized by the FDA in 2005 as monotherapy and then by the EMA in 2006 as add-on treatment to metformin and/or sulfonylurea where control was inadequate.
Parallel to the development of exenatide by Amylin Pharmaceuticals, Novo Nordisk worked on an alternative approach to designing a long acting GLP-1 receptor agonist. The GLP-1 peptide sequence was modified at the N-terminus to stabilise the molecule against degradation by DPP-4, and fatty acid side chains were added to enhance protein binding and prolong half-life. The length of the fatty acid chain was optimized for the balance between plasma half-life and GLP-1 receptor agonist potency [Knudsen et al. 2000]. The result was the molecule NN2211, named liraglutide, with one amino acid substitution from GLP-1 (Lys34Arg) and a 16-carbon palmitic acid side chain.
Liraglutide underwent phase III trials in the LEAD (Liraglutide Effect and Action in Diabetes) programme. As with exenatide, liraglutide was associated with a significantly greater weight loss compared with placebo in a dose-dependent manner. Liraglutide led to significant reductions in HbA1c over placebo in all the LEAD trials, amounting to between 1.2 and 1.6 HbA1c percentage points over 52 weeks [Blonde and Russell-Jones, 2009]. Liraglutide was tested at doses of 0.6, 1.2 and 1.8 mg. Weight loss was greatest in the higher 1.8 mg dose compared with lower doses across the LEAD studies. In terms of HbA1c reduction, whilst 1.8 mg outperformed the 1.2 mg dose when used as monotherapy (LEAD-3), no additional benefit of the higher dose was seen when used as add-on to oral hypoglycaemics (LEAD-1, -2 and -4). The 1.8 mg dose was, however, associated with an increased occurrence of predominantly gastro-intestinal side effects. Liraglutide became the second GLP-1 receptor agonist to be licensed for the treatment of type 2 diabetes, by the EMA in July 2009 and the FDA in January 2010. It was the subject of a NICE technology appraisal (TA203) in 2010 which recommended its use at a dose of 1.2 mg (but not 1.8 mg) for patients with T2DM and a BMI of ⩾35 kg/m2 [National Institute for Health and Clinical Excellence, 2010].
Mechanism of GLP-1 receptor agonist-induced weight loss
Endogenous GLP-1 is secreted by the L-cells of the intestine and acts via its G-protein linked membrane-bound receptor (GLP-1R). GLP-1R is expressed predominantly in the upper gastrointestinal tract, pancreatic islets [Bullock et al. 1996] and enteric visceral afferent nerves [Vahl et al. 2007]. In the central nervous system, GLP-1 is secreted as a neurotransmitter by neurons in areas of the brain including the hypothalamus. The GLP-1R is widely distributed throughout the brain [Merchenthaler et al. 1999] and densely expressed in hypothalamic nuclei involved in the regulation of appetite such as the paraventricular and arcuate nuclei [Shughrue et al. 1996].
GLP-1 has an appetite suppressant effect and the GLP-1 response to oral glucose is obtunded in obese patients [Færch et al. 2015]. The mechanism through which GLP-1 regulates appetite appears to be mediated through both peripheral and central nervous system (CNS) pathways. The action of GLP-1 on gut motility has been reviewed elsewhere [Marathe et al. 2011. An important effect is the slowing of gastric emptying which occurs as a result of GLP-1 activity [Delgado-Aros et al. 2002]. The resultant gastric stretch stimulates vagal afferent signals to the solitary nucleus of the medulla and onto the appetite centres of the hypothalamus to induce satiety or the area postrema to induce nausea. Early satiety and nausea are common side-effects of GLP-1 agonist drugs but are generally transient. Indeed, the retardation of gastric emptying by GLP-1 is subject to a rapid tachyphylaxis in experimental studies [Nauck et al. 2011]. Despite this, weight loss with GLP-1 agonists has been maintained in the absence of upper gastrointestinal side effects in clinical trials [Shyangdan et al. 2011], suggesting an alternative appetite suppressant pathway.
The CNS effects of GLP-1 have also been recently reviewed [van Bloemendaal et al. 2014]. It is estimated that 10–15% of endogenous GLP-1 secreted by intestinal L cells may reach the systemic circulation [Holst et al. 2005] where it is found in concentrations of approximately 10–40 pmol/l [Orskov et al. 1996]. From there, endogenous GLP-1 may be able to cross the blood–brain barrier and directly activate GLP-1R in the CNS [Kastin et al. 2002], while more substantial evidence attests to the ability of liraglutide and other GLP-1 analogues to do so [Secher et al. 2014; Hunter and Holscher, 2012]. Liraglutide has been shown to directly stimulate POMC neurons and inhibit neuropeptide-Y and Agouti-related peptide neurons of the arcuate nucleus resulting in appetite suppression [Secher et al. 2014]. These actions may also be accompanied by effects on other areas of the brain such as the mesolimbic system resulting in diminished food-induced reward signals and hence reduced food-seeking behaviour [Dickson et al. 2012; Dossat et al. 2011]. The efficacy of GLP-1 receptor agonists in maintaining weight loss may be explained by an attenuation of the fall in the anorexigenic hormone leptin that accompanies weight loss [Iepsen et al. 2014].
It is unclear whether GLP-1 influences energy expenditure. Some rodent models have demonstrated increased thermogenesis with administration of GLP-1 [Osaka et al. 2005]. Human studies, however, have yielded conflicting results [Harder et al. 2004; Horowitz et al. 2012].
In summary, while GLP-1 may increase energy expenditure in pharmacological concentrations, its predominant influence on body weight is through suppression of energy intake through direct and indirect actions on the appetite and food-reward centres of the brain and through local gastrointestinal effects.
Liraglutide: weight loss effects in diabetes: the LEAD and DURATION-6 trials
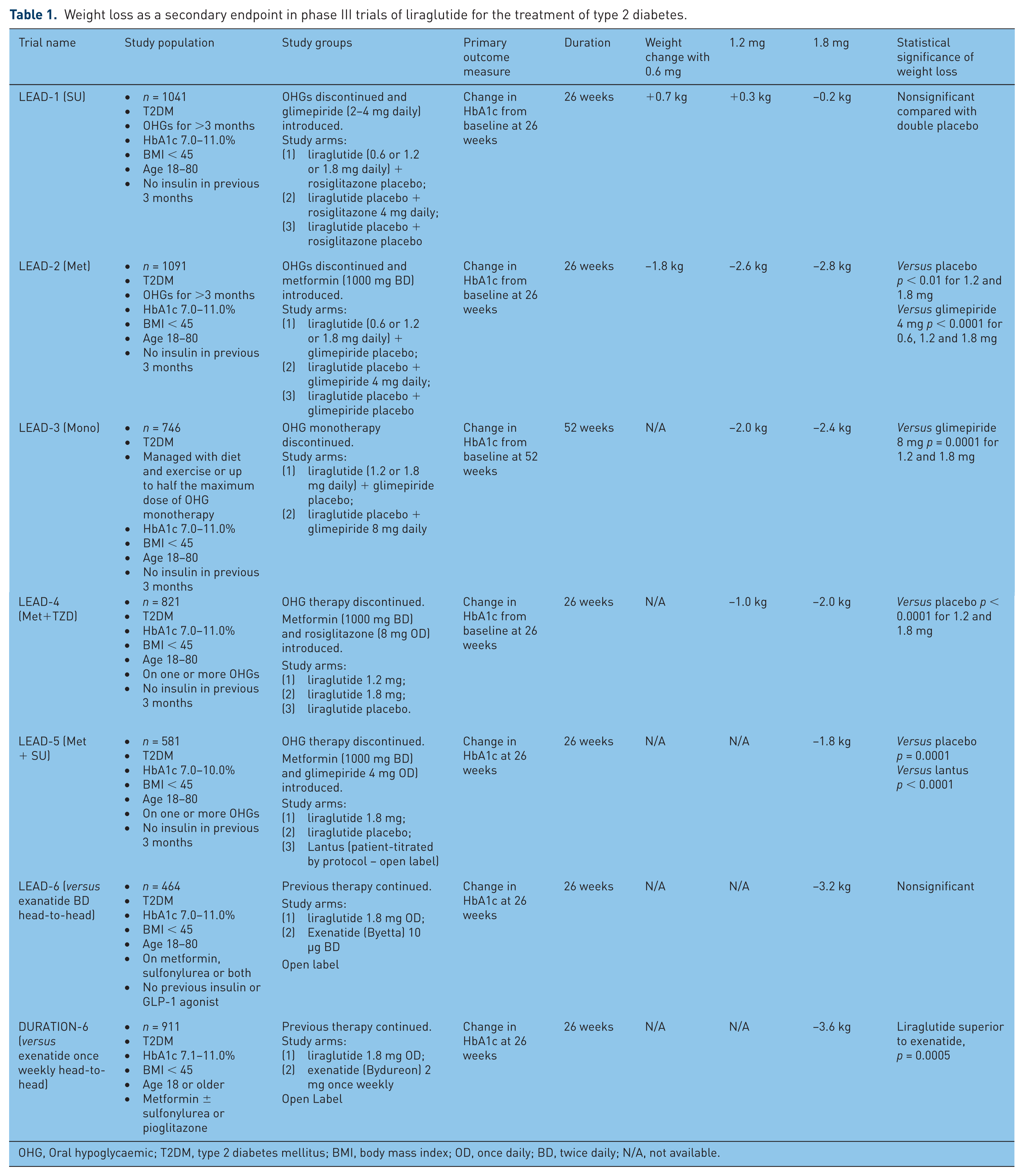
The LEAD and DURATION-6 trials are summarized in Table 1.
Weight loss as a secondary endpoint in phase III trials of liraglutide for the treatment of type 2 diabetes.
OHG, Oral hypoglycaemic; T2DM, type 2 diabetes mellitus; BMI, body mass index; OD, once daily; BD, twice daily; N/A, not available.
The 26-week, phase III, Liraglutide Effect and Action in Diabetes (LEAD) series of trials in patients with type 2 diabetes were powered to assess reduction in HbA1c as a primary endpoint, but also had sufficient statistical power to detect a 3% fall in weight. The results of LEAD trials 1 to 5 have previously been summarized by Blonde and Russell-Jones [Blonde and Russell-Jones, 2009].
In LEAD 1 (addition of liraglutide, rosiglitazone or placebo to sulfonylurea); participants in the active arm with a mean BMI of 30 kg/m2 did not lose significant weight compared with placebo [Marre et al. 2009].
In LEAD 2 (addition of liraglutide, glimepiride or placebo to metformin), participants with a mean BMI of 31 kg/m2 lost 2.8 kg at 1.8 mg dose over 26 weeks, significantly greater than the placebo arm (1.5 kg) (p < 0.0001) [Nauck et al. 2009].
In LEAD 3 (liraglutide versus glimepiride as monotherapy), participants with a mean BMI of 33 kg/m2 lost between 2.0 (1.2 mg dose, p < 0.0001) and 2.4 kg (1.8 mg dose, p < 0.0001), compared with a weight gain of 1.2 kg in the glimepiride arm [Garber et al. 2009].
In LEAD 4 (liraglutide or placebo added to combination of metformin and rosiglitazone), participants with a mean BMI of 33–34 kg/m2 lost 1.0 kg (1.2 mg dose, p < 0.0001) to 2.0 kg (1.8 mg dose, p < 0.0001) which was significantly different to the weight gain of 0.6 kg in the placebo arm [Zinman et al. 2009].
In LEAD 5 [addition of liraglutide (fixed-dose, 1.8 mg), insulin glargine (patient-titrated) or placebo to metformin and sulfonylurea], participants with a mean BMI of 30–31 kg/m2 lost 1.8 kg, significantly greater than placebo (−0.4 kg, p = 0.0001) and insulin glargine (+1.6 kg, p < 0.0001) [Russell-Jones et al. 2009].
Liraglutide was compared to exenatide in a head-to-head trial, LEAD-6 [Buse et al. 2009]. Patients on metformin, sulfonylurea or both were given either 1.8 mg liraglutide or 10 µg twice daily exenatide. After 26 weeks, participants in the liraglutide arm lost 3.24 kg compared with 2.87 kg in the exenatide arm, a nonsignificant difference (p = 0.2235), from a baseline BMI of 33 kg/m2. A degree of weight loss was seen in 78% of participants in the liraglutide arm and 76% on exenatide.
The DURATION-6 trial compared 1.8 mg liraglutide daily with 2 mg sustained-release exenatide (SR exenatide or Bydureon®) once weekly in patients with type 2 diabetes inadequately controlled on oral hypoglycaemic agents [Buse et al. 2013]. Mean BMI was 32.3 kg/m2 in both groups. Weight loss after 26 weeks (as a secondary endpoint) was significantly greater in the liraglutide arm (−3.57 liraglutide versus −2.68 kg SR exenatide, p = 0.0005).
Tolerability and safety of liraglutide in these trials was generally good. The most frequent side effects were gastrointestinal, predominantly nausea, which was most prevalent in the first few weeks then dissipated over time. The withdrawal rate in the liraglutide treatment arms was equivalent to or less than the comparator arms in all of the LEAD trials.
Liraglutide: weight loss effects in obesity (the SCALE trials)
Table 2 provides a summary of the SCALE trials. The first study to assess the effect of liraglutide on body weight and tolerability in obese individuals without type 2 diabetes was a double blind, placebo-controlled 20-week trial with open-label orlistat as a comparator and four different doses of liraglutide (1.2, 1.8, 2.4 and 3.0 mg) [Astrup et al. 2009]. All individuals received a 500 kcal per day energy-deficit diet and increased their activity throughout the trial which had an 84-week open-label extension that followed. It became clear from this dose-finding study that the 3 mg dose of liraglutide was the most effective dose for inducing weight loss at 20 weeks and beyond, and this dose was subsequently used for large scale phase III clinical trials. Mean weight loss after 20 weeks was 4.8 kg (1.2 mg dose), 5.5 kg (1.8 mg), 6.3 kg (2.4 mg) and 7.2 kg (3.0 mg). Orlistat resulted in a mean weight loss of 4.1 kg and placebo 2.1 kg.
Weight loss effect of liraglutide in the SCALE series.

OHG, Oral hypoglycaemic; T2DM, type 2 diabetes mellitus; BMI, body mass index; CPAP, continuous positive airway pressure; N/A, not available.
The weight loss efficacy of the 3 mg dose of liraglutide in patients with and without diabetes has been the subject of the Satiety and Clinical Adiposity – Liraglutide Evidence (SCALE) series of four trials [Novo Nordisk, 2015]. The series comprised trials in four patient categories: (1) overweight/obesity and type 2 diabetes; (2) overweight/obesity and prediabetes; (3) the SCALE - Maintenance trial which assessed the ability of high-dose liraglutide to maintain weight lost following a low-calorie diet and exercise intervention; and (4) obesity and sleep apnoea. All trials tested the effect of liraglutide on weight loss as an adjunct to diet and physical exercise.
SCALE - Diabetes trial
The results of the SCALE - Diabetes trial were presented at the American Diabetes Association conference in 2014 [Davies et al. 2014]. A total of 846 participants with type 2 diabetes, BMI ⩾27 kg/m2 and HbA1c of 7.0–10.0% on up to three oral agents were randomised to receive liraglutide 3.0 mg, liraglutide 1.8 mg or placebo plus lifestyle intervention for 56 weeks. Participants had a mean BMI of 37 kg/m2 and body weight of 106 kg. After 56 weeks, both the 3.0 and 1.8 mg doses of liraglutide performed significantly better than placebo, with percentage body weight losses of 5.9%, 4.6% and 2.0%, respectively. A total of 49.9% of individuals on 3.0 mg liraglutide lost ⩾5.0% of body weight, compared with 35.6% at the lower dose and 13.8% on placebo. A total of 23.4%, 14.4% and 4.3% lost >10% body weight in the three arms, respectively. The reported incidence of nausea was similar in the 3.0 and 1.8 mg arms, peaking at 16% after 4 weeks and subsiding thereafter.
SCALE - Obesity and Pre-diabetes trial
The results of the SCALE - Obesity and Pre-diabetes trial were published in July 2015 [Pi-Sunyer et al. 2015]. Patients with type 2 diabetes were excluded from this trial. Participants had either no diabetes or prediabetes as defined by the American Diabetes Association 2010 criteria [American Diabetes Association, 2010]. A total of 3731 participants with BMI ⩾30 kg/m2 or BMI ⩾27 kg/m2 and a cardiovascular comorbidity were randomized 2:1 to receive 3.0 mg liraglutide or placebo for a period of 56 weeks. In those without prediabetes (n = 1446), mean baseline body weight was 104 kg and BMI 37.5 kg/m2. Participants with prediabetes (n = 2285) were slightly more obese with baseline weight 108 kg and BMI of 39 kg/m2.
Weight loss achieved by those with and without prediabetes was similar and equated to a mean weight loss of −8.0% (8.4 kg) from a baseline of 106 kg, significantly greater than that in the placebo group (−2.6% or 2.8 kg). A total of 64% of subjects in the liraglutide arm achieved ⩾5% body weight loss compared with 27% in the placebo arm. A total of 33% of subjects achieved more than 10% body weight loss compared with 10% in the placebo arm. Liraglutide was associated with a reduced progression to prediabetes (7.2% against 20.7%) and increased reversal of prediabetes (69.2% against 32.7%) as defined by American Diabetes Association (ADA) criteria.
SCALE - Maintenance trial
The SCALE - Maintenance trial results were published in 2013 [Wadden et al. 2013]. In this study, obese (or overweight with BMI ⩾27 with comorbidity) subjects undertook a 1200–1400 kcal/day diet and exercise programme during a 12-week run-in. Patients were entered into the trial if they managed to lose ⩾5% body weight at 12 weeks prior to randomization. A total of 422 participants were randomized to receive 3.0 mg liraglutide or placebo for a period of 56 weeks. Primary endpoint analyses were percentage weight change from randomisation, proportion maintaining the initial 5% weight loss and proportion achieving an additional 5% weight loss from randomization. The mean weight loss at randomization was −6.0% from an initial baseline of 105–107 kg (BMI 38 kg/m2). Significantly more additional weight was lost by those on liraglutide (−6.2%) than those on placebo (−0.2%). A total of 81.4% in the liraglutide arm maintained a >5% loss from baseline weight, 48.9% on placebo. An extra 5% weight loss was achieved by 50.5% versus 21.8% of participants in the liraglutide and placebo arms, respectively, >10% weight loss by 26.1% and 6.3%, respectively.
SCALE - Sleep Apnoea
This was the smallest of the four trials with 359 patients [Collier et al. 2014]. Patients with moderate or severe obstructive sleep apnoea (OSA) were randomized to liraglutide or placebo for 32 weeks and reduction in the apnoea–hypopnea index (AHI) was evaluated at the end of the trial. Results showed a significantly greater reduction in weight and reduction in AHI in the liraglutide arm compared to placebo, suggesting improvement of OSA on liraglutide treatment.
Additional beneficial effects
A consistent if moderate improvement in low-density lipoprotein (LDL), very low-density lipoprotein (VLDL), total cholesterol and triglyceride concentrations was seen across the SCALE trials, alongside a small reduction in both systolic and diastolic blood pressure. Urine albumin:creatinine ratio in SCALE - Diabetes was reduced by a mean average of 18.4% in the liraglutide 3.0 mg arm against −2.3% with placebo.
Adverse events
Adverse events across the trials occurred more frequently in the liraglutide arms than placebo with the expected gastrointestinal events predominating. Withdrawal due to adverse events ranged between 8.5% in SCALE – Maintenance (in which the placebo arm saw 8.6% withdraw for the same reason) to 9.9% in SCALE – Obesity and Pre-diabetes (versus 3.8% with placebo).
The association of GLP-1 agonists and DPP-4 antagonists with pancreatitis has been the source of much controversy [Butler et al. 2013; Nauck, 2013]. One putative mechanism involves a GLP-1-induced increase in pancreatic duct turnover and metaplasia [Matveyenko et al. 2009]. Recent meta-analyses [Li et al. 2014; Giorda et al. 2015; Wang et al. 2015] have failed to show increased risk of acute pancreatitis with incretin therapies. In the SCALE - Obesity and Pre-diabetes trial, 10 patients on liraglutide and 1 on placebo developed acute pancreatitis (an event rate of 0.4 per 100 patient-years of exposure against 0.1 on account of the 2:1 randomization). There were no pancreatitis episodes in the other three trials. Lipase activity monitoring from the SCALE - Diabetes trial showed an early increase in activity which was sustained throughout the trial in those on liraglutide which was not seen with placebo. This did not rise above the upper limit of normal and no pancreatitis episodes were reported. Gallbladder-related complications, including cholelithiasis and cholecystitis, were more common in the liraglutide arm of each of these trials but serious adverse events involving gallbladder disease were rare.
Concern regarding the effect of liraglutide on thyroid C cells and risk of medullary thyroid cancer stems from evidence that rodents exposed to long-term liraglutide have an increased incidence of thyroid C-cell hyperplasia and medullary thyroid cancer [Bjerre Knudsen et al. 2010]. As a result, liraglutide’s drug information leaflet in the United States contains a black-box warning to this effect. In one large human study, no evidence of an increase in circulating calcitonin in association with liraglutide use was found [Hegedus et al. 2011]. Calcitonin concentrations were monitored during the SCALE trials. A small number of patients were found to have persistent elevations of calcitonin across the trials, more frequently in the liraglutide arms, but overall no statistical association between liraglutide and elevated calcitonin was seen. No incidents of medullary thyroid cancer were reported. The FDA has mandated the keeping of a 15-year medullary thyroid cancer registry for those prescribed 3.0 mg liraglutide.
Liraglutide treatment was associated with a small increase in mean heart rate of between two and four beats per minute across the four trials, although adverse event reports of palpitations and tachycardia occurred in less than 1% of participants in the treatment arms of each trial and were not significantly more frequent than for placebo. The increase in pulse rate returned to normal rapidly on cessation of treatment. Whether or not this heart rate increase is clinically meaningful will be addressed by currently ongoing post-marketing cardiovascular outcomes trials.
In summary, these trials point towards a moderate weight loss effect of liraglutide without evidence of severe toxicity and good tolerability.
Regulatory guidelines for obesity drug approval
Both the FDA and EMA have guideline thresholds for efficacy for any drug seeking marketing authorization as a weight-loss agent. Both agencies require weight loss to be the primary endpoint of the trial. The FDA efficacy benchmarks are (after one year) either (1) a placebo-subtracted mean weight loss of 5% body weight with statistically significant difference from placebo or (2) a proportion of patients in the active treatment group achieving 5% body weight loss of ⩾35% and significantly higher than placebo [Food and Drug Administration, 2007]. The EMA target (after 1 year) is of a mean weight loss of ⩾10% which in turn is ⩾5% more than placebo. If <5% more than placebo, the proportion of each group who respond can be considered as an alternative qualification, although a specified target for this difference is not defined [European Medicines Agency, 2007].
In composite data analysis of the SCALE trials, 3.0 mg liraglutide led to a 7.5% weight loss over 1 year compared with 2.3% for placebo which, although not quite meeting the EMA target, was considered to be sufficiently beneficial for approval [European Medicines Agency, 2015a]. The summary of evidence used by the Committee for Medicinal Products for Human Use in forming a favourable opinion of liraglutide for weight loss was published in February 2015 [European Medicines Agency, 2015b]. The FDA briefing document providing the basis for its approval was published in September 2014 [Food and Drug Administration, 2014].
How does liraglutide 3.0 mg compare with other new weight-loss pharmaceuticals
A comparison of liraglutide with other new weight-loss pharmaceuticals is given in Table 3. The efficacy of phentermine/topiramate was evaluated in three phase III clinical trials (EQUIP, CONQUER and SEQUEL). The EQUIP trial [Allison et al. 2012] included patients without significant obesity-associated comorbidities. Subjects received a dose of 15/92, 3.75/23 mg or placebo. After 56 weeks, mean weight loss was 10.9%, 5.1% and 1.6%, respectively. The CONQUER trial [Gadde et al. 2011] enrolled patients with obesity-related comorbidities: hypertension, prediabetes or type 2 diabetes on diet control or metformin. Three arms received 15/92, 7.5/46 mg or placebo and had a mean weight loss of 9.8%, 7.8% and 1.2%, respectively. A total of 78% of trial completers elected to continue on their randomized arms for a 52-week extension study, SEQUEL [Garvey et al. 2012]. They finished the study with mean weight losses of 10.5%, 9.3% and 1.8%.
Phase III trial weight loss efficacy of new weight loss drugs.

T2DM, type 2 diabetes mellitus; OD, once daily; BD, twice daily.
Lorcaserin was also the subject of three phase III trials: BLOOM (10 mg BD versus placebo), BLOSSOM (10 mg BD, OD or placebo) and BLOOM-DM (patients with type 2 diabetes, 10 mg BD, OD or placebo). In the BLOOM trial [Smith et al. 2010], patients in the lorcaserin arm lost a mean of 5.81% body weight compared with 2.16% on placebo after 1 year. At the end of 1 year, the active treatment arm was re-randomized to continue on lorcaserin or receive placebo for a further year. Both groups saw weight regain, the lorcaserin arm regained significantly less weight than the placebo arm. In the BLOSSOM trial [Fidler et al. 2011], 10 mg lorcaserin BD led to significantly more weight loss (5.8%) than 10 mg OD (4.7%) and placebo (2.8%). The same dose–response effect was not seen in the BLOOM-DM study [O’Neill et al. 2012]: participants on 10 mg BD lost and equivalent mean weight (4.5%) to those on OD (5.0%), both significantly greater than placebo (1.5%).
Four phase III trials were undertaken assessing naltrexone/bupropion, named: COR-I, COR-II, COR-BMOD and COR-DM. COR-I [Greenway et al. 2010] compared a dose of 16/360 mg with a dose of 32/360 mg and placebo. Both active treatment arms outperformed placebo but no significant dose–response effect of naltrexone was shown. COR-II [Apovian et al. 2013] examined the efficacy of the higher 32/360 mg dose against placebo with a placebo subtracted mean weight loss of 5.2% after 52 weeks (6.4% and 1.2%), with 50.5% versus 17.1% achieving ⩾5% weight loss at 1 year. COR-BMOD [Wadden et al. 2010] demonstrated that naltrexone/bupropion could significantly improve the weight loss achieved with an intensive behaviour modification intervention. Participants in COR-DM [Hollander et al. 2013] with type 2 diabetes, half of whom were treated with a sulfonylurea and a third with a thiazolidinedione, had a lesser weight loss than in COR-I and COR-II but which remained statistically higher than placebo (5.0% versus 1.8%).
Thus, in phase III pharmaceutical industry trials, 3.0 mg liraglutide resulted in slightly greater weight loss than naltrexone/bupropion and lorcaserin, and somewhat less weight loss than phentermine/topiramate. All of the newer agents’ weight loss effect was greater than that commonly observed with orlistat, in the region of 3% [Padwal et al. 2003] (Table 3).
Conclusion
For decades the world has lacked effective pharmacotherapy for the treatment of obesity. The few available drugs have been withdrawn over time due to unfavourable side-effect profiles. In Europe, with the exception of orlistat, there has been a void in the pharmacotherapy armamentarium, with few weight-reducing options available between diet and exercise and bariatric surgery. The SCALE clinical programme has shown that liraglutide, via manipulation of the physiological satiety hormone GLP-1, is an effective and well-tolerated anti-obesity drug with additional beneficial effects on diabetes, prediabetes and dyslipidaemia. It is a welcome addition to the treatment options available to the obesity physician. Further research studies will be useful to determine which patients are likely to derive the greatest benefit from high-dose liraglutide and to further define its place in the management of obesity.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare that there is no conflict of interest.