
Abstract
Prefibrotic primary myelofibrosis (pre-PMF) is a subtype of primary myelofibrosis, having only been formally defined as a distinct entity since 2016. Diagnosis, using clinical assessment, peripheral blood analysis, genetic and molecular analysis, and bone marrow biopsy, is needed to distinguish pre-PMF from other myeloproliferative neoplasms, in particular essential thrombocythemia. While the 2022 International Consensus Criteria and World Health Organization diagnostic criteria are used in current clinical practice to aid diagnosis, prognostication for pre-PMF remains challenging. Advances in molecular testing and cytogenetics may enable new risk stratification and prognostic tools to inform treatment decisions for pre-MF. In this review, we aim to give an overview on pre-PMF including the latest updates on diagnostics and prognostic indicators. Furthermore, due to limited reviews on pre-PMF, there still remains no standardized treatment pathway; thus, we summarize the current management advice as well as suggest further trials and areas of research that may improve care in pre-PMF patients.
Plain Language Summary
Prefibrotic primary myelofibrosis (pre-PMF) is a subtype of myelofibrosis which has only recently been defined as a distinct condition in recent years. However, despite advances in diagnostic techniques and development of new genetic screening tests, the overall identification and management of this condition remain challenging. In this review article, we aim to give an overview on all the recent developments in the diagnosis, prognosis and also potential treatment pathway for patients diagnosed with pre-PMF. Finally, we discuss future trials and areas of research that could improve care for patients with this condition.
Introduction
Prefibrotic primary myelofibrosis (pre-PMF) is a subtype of primary myelofibrosis (PMF) and a distinct myeloproliferative neoplasm (MPN) with specific histopathologic and clinical features. The concept of pre-PMF has evolved significantly over decades, gaining formal recognition as a distinct entity in the 2016 World Health Organization (WHO) classification. There are many challenging aspects in managing pre-PMF. The main challenge lies in establishing a diagnosis. Clinical features vary widely; some patients remain asymptomatic without needing any treatment, while others may progress rapidly. In addition, many clinical features overlap between pre-PMF and other MPN conditions, particularly essential thrombocythemia (ET). Ultimately, expert analysis of bone marrow biopsy is of paramount importance. Most of the prognostic scores available have been developed for overt PMF, and many of them may not apply for pre-PMF patients. The International Prognostic Score of thrombosis in Essential Thrombocythemia Thrombosis (IPSET), developed for assessing thrombotic risk in ET patients is the only prognostic score that has been validated for assessing thrombotic risk in pre-PMF.
Due to differences in prognosis and disease course, accurate diagnosis and distinction from ET and overt PMF are necessary. There is no standard of care for the management of patients with pre-PMF, as this cohort of patients is under-represented in clinical trials and research. This review aims to give an overview of the diagnosis, prognostication, and management of pre-PMF to help guide clinicians given the limited reviews on this condition.
Diagnosis
Clinical assessment, genetic and molecular analysis, and, most importantly, bone marrow histology are required to diagnose pre-PMF. 1 The challenges associated with diagnosing pre-PMF centers around differentiating it from other myeloid neoplasms, particularly ET and overt PMF, but also polycythemia vera (PV) and other MPNs.1,2 This can be difficult as the common driver MPN mutations (JAK2V617F, MPL, and CALR) can be seen in other MPN disorders, including pre-PMF, and there is no specific genetic mutation or blood test that will specifically distinguish pre-PMF. Overall distinction relies on clinical and histological features. 2
Clinical presentation
Symptoms of pre-PMF vary; some patients may be asymptomatic, while others experience constitutional symptoms, including weight loss, night sweats, itching, and abdominal pain.
Splenomegaly is a sign of extramedullary hematopoiesis. Clinically, those with pre-PMF tend to have larger spleen volume compared to ET patients1–3 and smaller to PMF. 4 In addition, splenomegaly is a more common finding in pre-PMF compared to ET. 2 A study found a larger median spleen size in PMF patients compared to pre-PMF patients, 20.8 and 17.95 cm respectively. 4
Patients with pre-PMF typically present with fewer and milder constitutional symptoms than those with overt MF (both primary and secondary).3,5 There are no available data on the proportion of asymptomatic patients with prefibrotic myelofibrosis. This is a suggested area of research, as it would help guide management for pre-PMF. We will discuss management separately, but approach to treatment is guided by symptoms, so it is vital to have more detailed data on this. It is recommended to use validated scores for evaluation of symptoms, such as MPN-10, to assess symptom burden and monitor progression over time. 1
International Consensus Criteria and WHO 2022 diagnostic criteria
The two diagnostic criteria that are used to diagnose pre-PMF are the International Consensus Criteria (ICC) and the WHO.1,6 There is much overlap between the two, with both requiring all three major criteria plus one minor criteria to be met for a diagnosis of pre-PMF to be made. Both are summarized in Table 1 and explored in further detail later.

Morphology of megakaryocytes in pre-PMF usually demonstrate a higher degree of megakaryocytic atypia than in any other MPN subtype; distinctive features of megakaryocytes include small to giant megakaryocytes with a prevalence of severe maturation defects (cloud-like, hypolobulated, and hyperchromatic nuclei) and presence of abnormal large dense clusters (mostly >6 megakaryocytes lying strictly adjacent).
It is recommended to use highly sensitive assays for JAK2V617F (sensitivity level <1%) and CALR and MPL (sensitivity level 1% to 3%); in negative cases, consider searching for noncanonical JAK2 and MPL mutations.
Assessed by cytogenetics or sensitive NGS techniques; detection of mutations associated with myeloid neoplasms (e.g., ASXL1, EZH2, IDH1, IDH2, SF3B1, SRSF2, and TET2 mutations) supports the clonal nature of the disease.
Minimal reticulin fibrosis (grade 1) secondary to infection, autoimmune disorder or other chronic inflammatory conditions, hairy cell leukemia or another lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies.
BM, Bone Marrow; CML, Chronic Myeloid Leukemia; ET, essential thrombocythemia; MPN, myeloproliferative neoplasm; pre-PMF, prefibrotic primary myelofibrosis; PV, polycythemia vera.
Laboratory investigations
Blood tests may show thrombocytosis, anemia, leukocytosis, raised lactate dehydrogenase (LDH) or cytopenias. 7 Both the ICC 2022 and WHO fifth edition criteria include a white blood cell (WBC) of >11 × 109/L as a minor criterion for pre-PMF. However, it is generally accepted that there are two phenotypes according to peripheral blood counts, a proliferative type and a dysplastic type with cytopenias. The cytopenic phenotype of pre-PMF is recognized as having at least one of the following: (1) white cell count <4 × 109/L, (2) hemoglobin <110 g/L for males and <100 g/L for females, and (3) platelets <100 × 109/L. This phenotype has a prognostic implication with an overall inferior outcome compared to the proliferative phenotype. 8 However, as discussed previously, it can be challenging to differentiate pre-PMF from ET, and this is discussed in further detail later.
Molecular and cytogenetics (including driver mutations)
Workup for pre-PMF needs molecular and genetic tests, and it is essential to examine for the presence of any of the MPN driver mutations; JAK2, CALR, and MPL.
Mutations in JAK2 are the most commonly found mutation in pre-PMF with a prevalence between 50% and 70%. 3 In pre-PMF, mean variant allele frequency (VAF) levels were shown to be lower compared to those with overt MF, while levels were higher compared to ET.3,7 Furthermore, similar to other MPNs, JAK2V617F mutated pre-PMF has been associated with a higher spleen volume and increased risk of thrombosis, in particular in the splanchnic veins. 9 Interestingly, splanchnic vein thrombosis was associated with lower VAF frequency (<10%). 10 If anything, this highlights the need to interpreting these data with the patient’s overall clinical condition in mind.
Following JAK2V617F mutations, CALR is the next most common mutation. This was found to be more common in pre-PMF than in ET, with 35.8% and 17.5% of patients, respectively. 7 Some evidence suggests that CALR type 1 mutation was more common in pre-PMF patients compared to type 2; however, this is not statistically significant and requires more research. It has been suggested that high platelet counts observed in pre-PMF patients may be linked to the higher frequency of CALR mutations compared to ET. However, the higher CALR mutation frequency in pre-PMF was not linked to higher incidence of leukemic transformation. 7 MPL is the least common mutation, found in only 2.0% of pre-PMF patients. 7 Little is known about the clinical presentation associated with MPL mutations for pre-PMF patients.
Pre-PMF patients who are triple negative for the MPN driver mutations are approximately 10%–20%. 2 The proportion of triple negative cases is higher in pre-PMF compared with overt MF. 11 Similar to overt MF, triple negative pre-PMF is clinically associated with anemia, transfusion dependency, and leukocytosis and importantly is also associated with a poor prognosis. 12
Overview of high molecular risk and unfavorable cytogenetics
In comparison to overt MF, pre-PMF is a less genetically unstable disease, with fewer high molecular risk (HMR) mutations and abnormal karyotype observed in this patient cohort.1,9 The presence of two or more HMR mutation confers poorer prognosis. 3 Kim et al. recently investigated gene alterations in 229 patients with PMF, secondary MF and pre-PMF, in particular looking at the age-related clonal hematopoiesis and Clonal Hematopoiesis of Indeterminate Potential (CHIP)-associated mutations. In this study, 211 patients of the study population were identified to have mutations. 13 They found that patients with pre-PMF had fewer mutations (1.65), compared to secondary MF (1.78) and PMF (1.91). Regarding the types of mutations present, in PMF, ASXL1 (35.2%) was the most common followed by TET2 (9%) and U2AF1 (6.6%), while for patients with pre-PMF the most common mutations were TET2 (17.5%) followed by SF3B1 (10%) and DNMT3A (10%). In terms of prognosis, this group found that ASXL1, U2AF1, SETBP1, and SRSF2 were associated with inferior overall survival in pre-PMF patients, while TP53 mutation was associated with poorer outcomes in PMF patients. 13 Notably, none of the pre-PMF patients had a very high risk karyotype, as defined by the Mutation-Enhanced International Prognostic Score System (MIPSS). 13
Mimics of pre-PMF
Essential thrombocythemia
There are limitations to the ICC and WHO 2022 histology-based diagnostic criteria, particularly in relation to distinguishing between pre-PMF and ET, with some studies questioning its reliability and reproducibility.11,14,15 In clinical practice, this still poses a diagnostic dilemma. Upon reviewing studies for MPN patients, it has been highlighted that when diagnoses were revisited, patients had been misdiagnosed with ET and were subsequently found to have pre-PMF. A multidisciplinary team approach is therefore needed, relying on clinical and histopathology expertise. The ability to differentiate between pre-PMF and ET, while challenging, is incredibly important as the rates of transformation to acute leukemia and overt MF are reported to be higher in pre-PMF compared to ET. The pre-PMF cumulative incidence of leukemia over 10 years is estimated to be 2.3% compared to 1.9% in ET.7,16
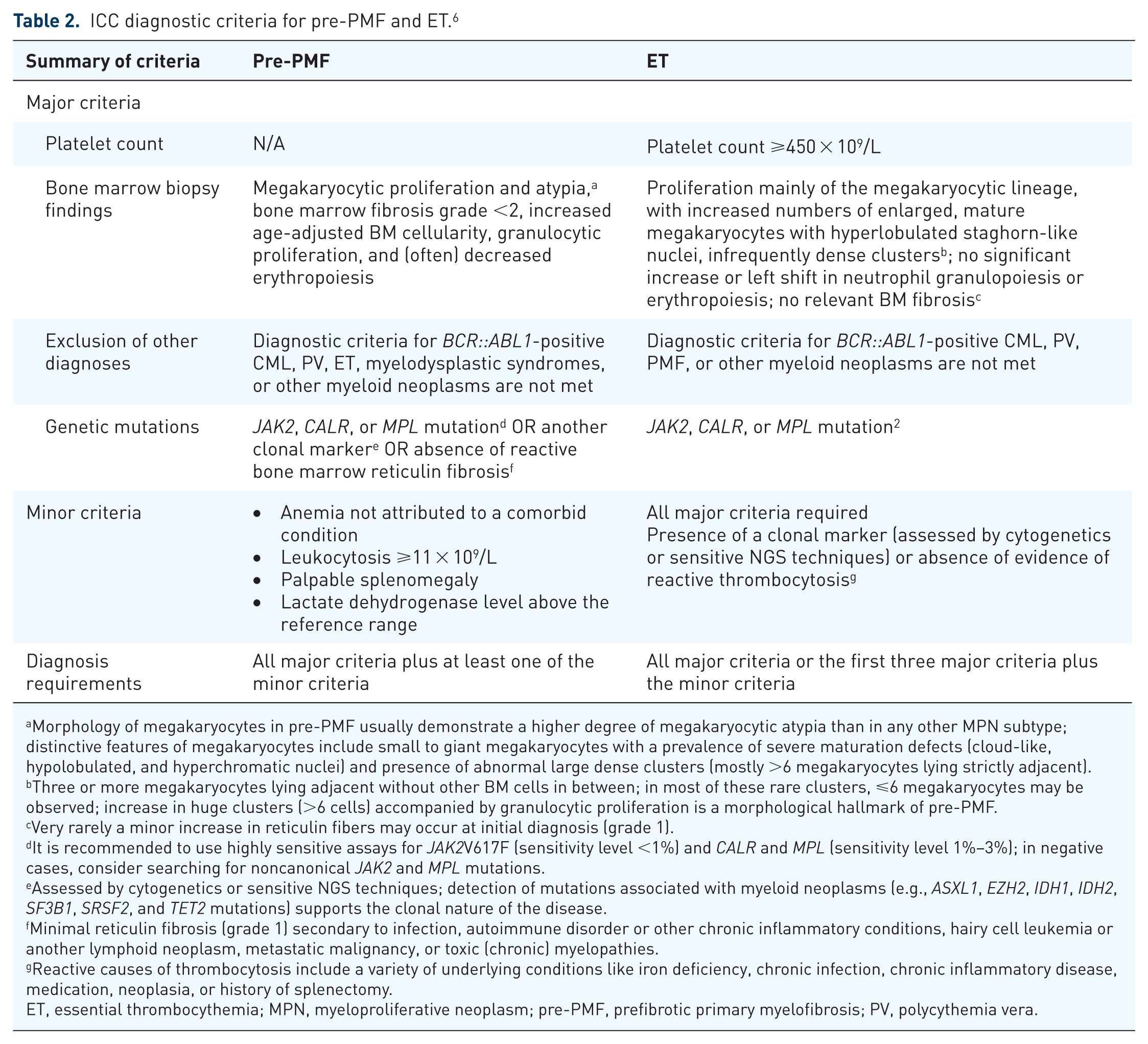
More recent research has found that with a systematic approach and sufficient supporting clinical data, there was reproducible consensus on bone marrow histological evaluation between histopathologists.17–19 The differences between a diagnosis of pre-PMF and ET according to the ICC of myeloid neoplasms are summarized in Table 2.
ICC diagnostic criteria for pre-PMF and ET. 6
Morphology of megakaryocytes in pre-PMF usually demonstrate a higher degree of megakaryocytic atypia than in any other MPN subtype; distinctive features of megakaryocytes include small to giant megakaryocytes with a prevalence of severe maturation defects (cloud-like, hypolobulated, and hyperchromatic nuclei) and presence of abnormal large dense clusters (mostly >6 megakaryocytes lying strictly adjacent).
Three or more megakaryocytes lying adjacent without other BM cells in between; in most of these rare clusters, ⩽6 megakaryocytes may be observed; increase in huge clusters (>6 cells) accompanied by granulocytic proliferation is a morphological hallmark of pre-PMF.
Very rarely a minor increase in reticulin fibers may occur at initial diagnosis (grade 1).
It is recommended to use highly sensitive assays for JAK2V617F (sensitivity level <1%) and CALR and MPL (sensitivity level 1%–3%); in negative cases, consider searching for noncanonical JAK2 and MPL mutations.
Assessed by cytogenetics or sensitive NGS techniques; detection of mutations associated with myeloid neoplasms (e.g., ASXL1, EZH2, IDH1, IDH2, SF3B1, SRSF2, and TET2 mutations) supports the clonal nature of the disease.
Minimal reticulin fibrosis (grade 1) secondary to infection, autoimmune disorder or other chronic inflammatory conditions, hairy cell leukemia or another lymphoid neoplasm, metastatic malignancy, or toxic (chronic) myelopathies.
Reactive causes of thrombocytosis include a variety of underlying conditions like iron deficiency, chronic infection, chronic inflammatory disease, medication, neoplasia, or history of splenectomy.
ET, essential thrombocythemia; MPN, myeloproliferative neoplasm; pre-PMF, prefibrotic primary myelofibrosis; PV, polycythemia vera.
The ICC diagnostic criteria highlight key bone marrow morphology differences between pre-PMF and ET. Increased BM cellularity is not associated with ET, whereas it is seen in pre-PMF. Bone marrow findings of patients with pre-PMF have more megakaryocytic atypia, including hypolobulation, hyperchromatic nuclei, and abnormally large dense clusters. 6 Pre-PMF also often has decreased erythropoiesis, typically with higher levels of immature erythroid precursors due to ineffective hematopoiesis.1,6
Another notable difference in the ICC diagnostic criteria between pre-PMF and ET was the pre-PMF minor criteria, where the presence of any of the four minor criteria can indicate a diagnosis of pre-PMF over ET. However, it is important to note that the minor criteria can be present in ET patients, with one study showing that 48% of ET patients exhibited one or more of the minor criteria, emphasizing the importance of a bone marrow biopsy. 20 Patients with pre-PMF tend to have a higher leukocyte count, platelet count, and LDH levels compared with patients with ET, as well as more frequently exhibiting splenomegaly. 7 It is noted that LDH is one of the most important factors of pre-PMF. 1 Pre-PMF does also tend to have higher numbers of circulating CD34-positive cells than ET with a median count of 6.6 × 109/L in pre-PMF compared to 3.6 × 109/L in ET. 7
There has been some work in developing diagnostic models that do not rely on molecular tests to make them more accessible in order to help discriminate between pre-PMF and ET. In 2012, Carobbio et al. designed the Bergamo algorithm, which was developed to be used in patients with an ET-like phenotype and utilized hemoglobin, white blood cell count, and LDH in a stepwise fashion to aid distinguishing between a diagnosis ET or pre-PMF, factoring in a margin of error of 10% at each step. 21 Another study built on this algorithm further to include splenomegaly and applied the other minor criteria in a logarithmic formula rather than a stepwise manner. 22
Lekovic et al. 23 formulated a pre-PMF predictive score calculator based on age over 60, raised LDH and splenomegaly. This model had a high predictive value for pre-PMF in patients thought to have ET, with those scoring ⩾2 points having positive predictive value of 69.8%, which then rose to 88.2% with scores ⩾3. 23 More recently, Barbui et al. developed a diagnostic model utilizing WBC count and platelet levels, noting higher WBC/platelet levels being associated with an increased probability of pre-PMF diagnosis, independent of LDH levels and splenomegaly. 24 Table 3 summarizes the differences between these diagnostic models.

ET, essential thrombocythemia; LDH, lactate dehydrogenase; pre-PMF, prefibrotic primary myelofibrosis; PPV, positive predictive value; WBC, white blood cell.
While these diagnostic models can aid in the diagnosis of pre-PMF, further validation in larger patient groups is required. Despite this, these models could help in future clinical practice to triage patients presenting with thrombocytosis that need urgent hematological evaluation and raise earlier suspicion of pre-PMF.
Regarding differences from a molecular perspective, as mentioned earlier, studies to date have not demonstrated a significant difference in the proportion of driver mutations between ET and pre-PMF patients. 7 However, as highlighted, there has been a notable difference observed in JAK2V617F VAF with pre-PMF patients having higher median levels compared to those with ET (46% vs 25% respectively). 25 Additionally, as mentioned, splenomegaly is more common in pre-PMF patients compared to ET. 2
Interestingly, in a cohort study of patients with thrombocytosis by Kuo et al., they found that patients with ET experienced a greater incidence of cerebral ischemic stroke, while those with pre-PMF were more likely to develop splanchnic vein thrombosis. 26 The relationship between pre-PMF and thrombosis will be discussed in further detail later.
Distinguishing between pre-PMF and ET is not always possible and at times only becomes apparent after the disease has developed further. It is likely that in the future with developments in genetic parameters and further relevant molecular markers, we may be able to decipher the bone marrow results with the clinical phenotype further. 1 Of interest, an artificial intelligence (AI) model was developed to distinguish between pre-PMF and ET, which reported high accuracy of diagnosis in 200 patients in Italy. 27 This is a novel field and, with further research, may provide new algorithms to overcome diagnostic challenges in MPNs, particularly for pre-PMF as highlighted above.
Overt myelofibrosis
The ICC and 2022 WHO diagnostic criteria for pre-PMF and overt MF are very similar, with the only difference being reticulin and/or collagen fibrosis grades 2 or 3 seen on bone marrow biopsy in overt MF compared to less than two in pre-PMF. 6 The two conditions are similar in a continuum of mutations, including ASXL1, EZH2, SRSF2, and IDH1/2, and cytogenetic abnormalities that have an influence on prognosis.1,9 However, as already mentioned, pre-PMF is associated with significantly fewer chromosomal abnormalities than overt MF, making it a less genetically unstable disease. 9 In particular, ASXL1 mutations have been found to be less frequently present in pre-PMF and the mean VAF lower in pre-PMF compared to overt MF, being 36.1% and 7.5%, respectively. 13
As its name suggests, pre-PMF is typically a milder condition than overt MF. Patients with pre-PMF are more frequently female and younger, and tend to have higher hemoglobin, leukocyte, and platelet counts.3,9,13 Consequently, pre-PMF is associated with fewer adverse risk factors compared to overt MF and so less associated with adverse clinical outcomes.9,13 This is to be taken into account when applying risk scores designed for overt MF patients to pre-PMF patients.
MPN, not otherwise specified or unclassifiable
MPN—not otherwise specified or unclassifiable (MPN-NOS, MPN-U)—can also be a challenge to distinguish from pre-PMF. One main reason for this is that low-grade fibrosis can also be seen in MPN-NOS and MPN-U, although bone marrow biopsy tends to be normocellular with evidence of myelodysplasia and myeloproliferation. By comparison, the bone marrow tends to be hypercellular with no dysplasia in pre-PMF. 2
Polycythemia vera
Due to the higher levels of hemoglobin and hematocrit, patients who are found to have low-grade fibrosis may be misdiagnosed as pre-PMF rather than PV. It is important to note that CALR and MPL mutations are nearly never present in PV, and conversely that a JAK2 mutation is nearly always present.2,28,29 Low levels of serum erythropoietin are seen in PV but not in pre-PMF; however, data from large trials looking at levels of erythropoietin levels in patients with pre-PMF are lacking, due to the reduced frequency of anemia observed in this cohort of patients.1,2
Autoimmune myelofibrosis
Autoimmune myelofibrosis (AIMF) is a benign cause of myelofibrosis that occurs with a known autoimmune disease in secondary AIMF, or in the absence of an overt autoimmune disease in primary AIMF. AIMF is associated with absent or minimal splenomegaly, unlike pre-PMF, and tends to have no eosinophilia or basophilia and normal morphology in all cell lineages. 30 While the main differentiating feature is a known history of an autoimmune condition, nearly all AIMF demonstrates lymphocytic infiltration with both CD3+ and CD20+ lymphocytes. Importantly, atypical megakaryocytes are generally absent in AIMF, while these are a major criterion in a pre-PMF diagnosis. Differentiating between AIMF and pre-PMF is important due to the benign nature of AIMF and its associated good prognosis, typically responding well to corticosteroids. 31
Risk stratification
Despite being recognized as a distinct disease for multiple decades with evidence of lower overall survival when compared to ET, there is still no risk stratification tool specifically for pre-PMF, and therefore, clinicians often rely on prognostic scoring tools for overt MF. 2 These initially included the IPSS, DIPPS, and then DIPSS-plus through progressive iterations. 5 There is a paucity of research on using these tools in pre-PMF, and therefore, clinicians must be wary of this when using these tools to guide clinical practice. 2
Following DIPSS-plus, MIPSS70 and then MIPSS70 plus were developed to include further molecular risk factors and distinguish between the different gene and karyotype risks. 2 Designed to aid prognostication of patients with PMF of transplantation age (under 70 years), this study group did include pre-PMF patients. MIPSS70 utilized the following factors to stratify patients into three risk categories: hemoglobin (<100 g/L), leukocyte count (>25 × 109/L), platelet level (<100 × 109/L), circulating blast count (>2%), bone marrow fibrosis (grade 2 or more), presence of constitutional symptoms, driver mutations (CALR type 2, JAK2V617F, MPLW515X), absence of CALR type 1-like mutation, HMR category (presence of a mutation in any of ASXL1, EZH2, SRSF2, IDH1/2), and two or more HMR mutations. 32 Each of these factors represented an independent predictor of survival.
Because of these updated risk stratification tools, and the inclusion of pre-PMF in the newer models, the Society of Hematologic Oncology do suggest using these tools (in particular DIPSS and MIPSS70). However, they also apply caution and remain conscious of their limitations, particularly in relation to their lack of validation in pre-PMF patients to aid management. 2
Given that not all centers can easily access genomic sequencing, Tefferi et al. developed the AAA model that utilized age as well as the full blood count to predict prognosis in ET patients. 33 Lucijanic et al. have recently reviewed its applicability in patients with pre-PMF and found that it can predict overall survival. 34 However, it should be noted that this work was limited by low numbers of patients as well as short follow-up time, so further large-scale studies are required to confirm the validity of the AAA model in this patient cohort.
Thrombosis risk
Thrombosis risk in pre-PMF is roughly double that of a person in the general population, and in fact similar to ET risk. 7 Thrombotic events have been found in one study to occur more frequently in the splanchnic area in pre-PMF patients. It is possible that young age combined with female sex and the JAK2V617F mutation may lead to the development of a prothrombotic state in the splanchnic veins. 13
Similar to above, no risk scores to predict bleeding or thrombotic episodes have been specifically created for pre-PMF. International Prognostic Score of thrombosis in Essential Thrombocythemia Thrombosis (IPSET-T) and revised IPSET-T utilizes age (over 60 years), prior thrombotic event, cardiovascular risk factors (one or more of hypertension, diabetes mellitus, active smoking), and presence of JAK2V617F mutation to determine thrombosis risk in patients with ET. The revised IPSET-T included the additional fourth risk group of very low risk in comparison to IPSET-T with three risk categories.35,36 While created specifically for patients with ET, IPSET-T, and less so revised IPSET-T, has been validated to determine thrombotic risk for patients with pre-PMF.36,37
Bleeding risk
Major bleeding appears to be more common in pre-PMF compared to ET, with an international study finding that the rate of major bleeding in pre-PMF was 1.39% compared to 0.79% in ET. 38 However, a smaller study comparing the clinical course of patients with pre-PMF and ET found no significant difference in major bleeding incidence, even when only patients deemed low risk and not receiving cytoreduction were analyzed. 7 Finazzi et al. 38 looked at studies comparing ET and pre-PMF, highlighting one study that showed the 10-year cumulative incidence of bleeding in pre-PMF to be as high as 19.6% compared to 14.9% in ET, although this was not statistically significant. Bleeding in pre-PMF was predicted by factors such as leukocytosis, previous hemorrhage and, importantly, the use of aspirin therapy, with low-dose aspirin exacerbating bleeding risk. Interestingly, by contrast to ET, thrombocytosis was not a risk factor for bleeding in pre-PMF patients. 38
Prognosis
Overall, patients with pre-PMF have been shown to have superior overall survival and leukemia-free survival compared to overt PMF, but inferior to patients with ET. 3 In terms of driver mutations, pre-PMF patients with CALR type 1 mutations have been associated with a more favorable outcome, similar to overt PMF. Notably, there was no statistically significant difference observed in survival rates between those with JAK2V617F, MPLW515, and CALR type 1 in pre-PMF patients, while triple negative disease was associated with poorer outcomes. 3 Patients with pre-PMF have a longer median overall survival compared to those with overt PMF, 14.7 and 7.2 years, respectively, with similar trends in leukemia-free survival. 1 Recently, a number of studies have demonstrated correlation between JAK2 VAF levels and several disease outcomes in MPN patients. Patients with PV, with higher VAF (>50%) being associated with increased risk of progression. 12 A study in Denmark also demonstrated a higher risk of severe coronary atherosclerotic disease in patients with a higher VAF (>52%) in MPN patients with PV, ET, and MF. 39 It is unclear if a similar risk exists for JAK2 positive pre-PMF patients.
As discussed earlier, HMR mutations are less frequently observed in pre-PMF compared to overt PMF patients. However, if present, these can be associated with poorer prognosis, with ⩾2 HMR mutations being associated with reduced survival (12.7 years with one HMR mutation vs 2.6 years with ⩾2 HMR mutations). 3
Risk of progression to overt PMF
In general, not all patients with pre-PMF transform to overt PMF and timing to progression remains unclear. One group reviewed patients with the whole spectrum of disease of pre-PMF versus ET based on the WHO 2016 criteria. 20 Cumulative risk of progression to PMF was 36.9% after both 10 and 15 years for pre-PMF patients, which then increased to 59.6% at 10 years for patients with a platelet count <450 × 109/L at diagnosis. The risk for ET patients was significantly lower (7.5% and 11.7% at 10 and 15 years, respectively). Barbui et al. demonstrated similar findings, with a lower 15-year cumulative risk of overt fibrotic progression observed in ET compared to pre-PMF patients (9% vs 17%, respectively). 40
Risk of progression to acute leukemia
As expected, risk of transformation to acute leukemia is generally higher in overt PMF compared to pre-PMF patients. Data are limited, but the largest study to date by Guglielmelli et al. reported a 5-year cumulative incidence of leukemia of 7% compared to 11% in overt PMF. 3 Risk factors associated with transformation to leukemia in pre-PMF patients have included age>65 years, leukocytosis (WBC >15 × 109/L), raised LDH (>1.5 × upper limit of normal), and cytogenetic abnormalities. 21
Treatment
Goals of treatment are focused on reducing thrombotic risk and disease modification, similar to the management of ET. 1 Risk stratification of pre-PMF patients can help to guide treatment; however, to date, there are currently no standardized guidelines for the management of pre-PMF. Care must be taken when applying principles of treatment similar to that of ET, as we have highlighted above, although there are overlapping clinical features, the risks of transformation and progression differ, and so treatment must be individualized according to the clinical picture.
Observation
It is often recommended that patients with low symptom burden and deemed low risk, identified using prognostic scores such as DIPSS or MIPSS70, should be observed only without active treatment. As this cohort of patients has a relatively favorable prognosis, there is currently no evidence to suggest that early treatment prevents disease progression. For patients in this cohort with risk factors for thrombosis, low-dose aspirin can be used to reduce thrombotic risk. 1
Cytoreduction
For leukocytosis or thrombocytosis, hydroxycarbamide is often used first-line as cytoreductive treatment. For younger patients with longer life expectancy, interferon alfa has shown evidence of controlling disease progression by reducing the JAK2V617F allele burden.1,41 There are ongoing trials looking at the use of ropeginterferon-alpha in pre-PMF with current data showing promising cytoreduction, significant molecular remission, reduction in allele frequency, and fibrosis improvement in pre-PMF. Reassuringly, no progression to overt PMF or blast phase was seen during the study. In addition, the good tolerability of this treatment makes it an appealing option. 41
Anagrelide is used less in practice due to concerns of increasing bone marrow fibrosis, as well as frequent cardiac side effects.2,42
JAK inhibitors can be used in selected cases to help control symptom burden or splenomegaly. A study of 130 pre-PMF patients by Masarova et al. found that the use of JAK2 inhibitors in symptomatic patients requiring treatment was associated with improved outcomes. 43 The data on the use of JAK inhibitors in pre-PMF is scarce, however, and there are no trials at present studying the use of JAK inhibitors for pre-PMF patients.
Anemia
Significant anemia is uncommon in pre-PMF and it is rare for patients to be transfusion dependent. 2 It is imperative in these cases to investigate other causes of anemia and re-evaluate the disease.
Thrombosis
Thromboses are generally treated with aspirin (if an arterial event) or anticoagulation (if a venous event) depending on the nature of the thrombotic event. 40 These decisions are balanced with bleeding risk. Assessment for cytoreduction is vital at the time of diagnosis of thrombosis as this is generally considered an indication to start cytoreductive treatment. 2
Transplant
Allogeneic stem cell transplant should be considered in patients who have high risk molecular features (based on MIPSS70), as well as signs of clonal evolution and disease progression. 2 However, data on outcomes of transplant in pre-PMF patients is limited.
Pre-PMF in pregnancy
There is little data regarding pregnancy in pre-PMF, with more data available for pregnancy in patients with ET. A study reviewing MPN databases in the University of Florence, Italy and the Mayo Clinic, USA identified pre-PMF phenotype as a risk factor for fetal loss, with 7 of the 16 (44%) pre-PMF patients experiencing fetal loss compared to none of the eight patients with PMF or post-ET MF experiencing fetal loss. 44 Although this was a small study of just 24 pregnancies, the majority were pre-PMF patients, and this highlights that management of such cases needs careful risk assessment and expert management with an MDT approach.
Psychological support
Finally, we must not underestimate the anxieties a patient may have regarding the diagnosis of pre-PMF. This can be driven by a number of factors, including lack of standardized treatment strategies, diagnostic challenge, and symptom burden.
Figure 1 summarizes a potential treatment pathway for patients diagnosed with pre-PMF.

Flowchart demonstrating an overview for the management of a patient with suspected pre-PMF.
Future directions
Trials
Enrolling pre-PMF patients in clinical trials should be considered, as this may lead to development of treatment algorithms specific to pre-PMF patients. There are ongoing trials looking at the role of ropeginterferon alfa-2b in pre-PMF with promising results. 45
JAK2 inhibitors are also being used in clinical practice to treat pre-PMF patients; however, data on the long-term outcomes of these patients are limited and highlight an ongoing need for large-scale prospective studies.
Diagnostic advances
As most of the large studies research mutation analysis of driver and selected non-driver genetic mutations, there may be other HMR mutations leading to negative outcomes in pre-PMF that are not yet known. There are promising advances in diagnostic methods that could improve this and uncover new mutations.
Role of AI
As AI is gaining popularity in diagnostic medicine, and this was incorporated into an algorithm to distinguish pre-PMF from ET patients, this will likely be a growing field in the future, in particular given the diagnostic challenges of pre-PMF.27,46
Conclusion
Pre-PMF remains a challenge given the overlap of clinical presentation and features with other MPNs, particularly ET. Our review summarizes recent studies in pre-PMF, where there has been particular focus on establishing the diagnostic criteria as well as developing prognostic models. However, much of the data have been extrapolated from ET and PMF, and there are limited clinical trials for pre-PMF patients. Future work is required to refine diagnostic and prognostic tools using novel technologies, as well as examine long-term outcomes in pre-PMF patients.