
Abstract
In haploidentical hematopoietic stem cell transplantation, preexisting donor-specific anti-HLA antibodies (DSAs) in recipients have been associated with graft rejection, poor graft function (PGF), and unfavorable clinical outcomes, with relatively well-established consensus strategies for its management. However, reports on the emergence of de novo DSAs after transplantation and the corresponding therapeutic approaches remain limited. Here, we describe a case of a 14-year-old patient with myelodysplastic syndrome who developed de novo DSAs as early as day 15 post-transplantation, with a peak mean fluorescence intensity of 12,280, which was associated with PGF. To reduce DSA levels, plasma exchange combined with intravenous immunoglobulin and rituximab was administered, followed by a series of interventions including mesenchymal stem cell infusion. These measures facilitated graft function recovery, controlled transplant-related complications, and ultimately led to successful engraftment.
Keywords
Introduction
Allogeneic hematopoietic stem cell transplantation (allo-HSCT) remains a curative therapeutic option for patients with high-risk hematologic malignancies. In recent years, haploidentical hematopoietic stem cell transplantation (haplo-HSCT) has been increasingly adopted across numerous centers, particularly in the context of lacking HLA-matched donors or the need for urgent transplantation, owing to substantial advances in the prevention of graft-versus-host disease (GVHD).1,2 In haplo-HSCT, preformed donor-specific anti-HLA antibodies (DSAs) present in recipients have been closely associated with graft failure and poor graft function (PGF), ultimately contributing to unfavorable post-transplant outcomes.3,4 However, reports on the development of de novo DSAs are scarce, and their impact on the outcomes of haplo-HSCT remains unclear. Here, we report a case of a patient with myelodysplastic syndrome (MDS) who developed de novo DSAs on day 15 following haplo-HSCT, with a mean fluorescence intensity (MFI) reaching 12,280, which may have contributed to primary PGF. After receiving a series of treatments, the patient ultimately achieved successful engraftment. The reporting of this case conforms to the CARE guidelines for clinical case reporting. 5
Case presentation
A 14-year-old girl presented to the Department of Hematology at the First Affiliated Hospital, Zhejiang University School of Medicine with a 4-month history of recurrent anemia. She initially experienced dizziness and fatigue without an obvious cause. On May 18, 2024, routine blood tests revealed a white blood cell (WBC) count of 2.65 × 109/L, hemoglobin of 81 g/L, and platelet count of 181 × 109/L. No improvement was observed after treatment with Leucogen tablets, a cysteine derivative that acts as a hematopoietic stimulator to support leukocyte recovery, and her hemoglobin level progressively declined. At Zhejiang Children’s Hospital, blood tests showed a hemoglobin level of 60 g/L. Bone marrow aspiration resulted in a dry tap, and biopsy revealed marrow fibrosis with immature megakaryocytic hyperplasia. Abdominal ultrasound showed no splenomegaly, and a definitive diagnosis could not be established. She was subsequently referred to the Children’s Hospital of Fudan University, where bone marrow aspiration again yielded poor material, with no blasts or abnormal cells detected. Whole-exome sequencing identified mutations in GATA2 (p.G268Efs*58, 49.5%), RASGRP2 (p.L518=, 46.9%), and KMT2B (c.7297+7G>T, 49.0%). A chromosomal microarray revealed a full mosaic loss of chromosome 7, and bone marrow biopsy confirmed reticulin fibrosis (+++). Her father tested negative for germline GATA2 mutations. Supportive care, including red blood cell transfusions, was provided at local hospitals. On September 10, 2024, she presented to our center for further evaluation. Laboratory tests showed WBC 1.8 × 109/L, neutrophils 0.34 × 109/L, hemoglobin 45 g/L, and platelets 89 × 109/L. Bone marrow evaluation indicated reduced erythroid and granulocytic precursors without blasts; flow cytometry revealed 1.93% myeloid blasts. Cytogenetic analysis demonstrated multiple clonal abnormalities: 45,XX,-7[5]/45,XX,sl,-3,+mar[2]/46,sl,+8[2]/46,XX[1]. Based on a comprehensive evaluation, she was diagnosed with myelodysplastic syndrome with low blasts (MDS-LB), classified as high-risk according to both the IPSS-R (5.5) and IPSS-M (1.33).6,7 Allo-HSCT was planned using her haploidentical father as the donor. HLA typing showed a 7/12 allele match (Table 1). Pre-transplant screening for class I/II anti-HLA antibodies was performed on July 24, 2024, approximately 2 months prior to transplantation, and was negative, with both class I and II antibody MFI values <100. The patient was blood type B Rh-negative, and the donor was B Rh-positive.
HLA typing results and de novo anti-HLA class I antibodies detected post-transplantation.
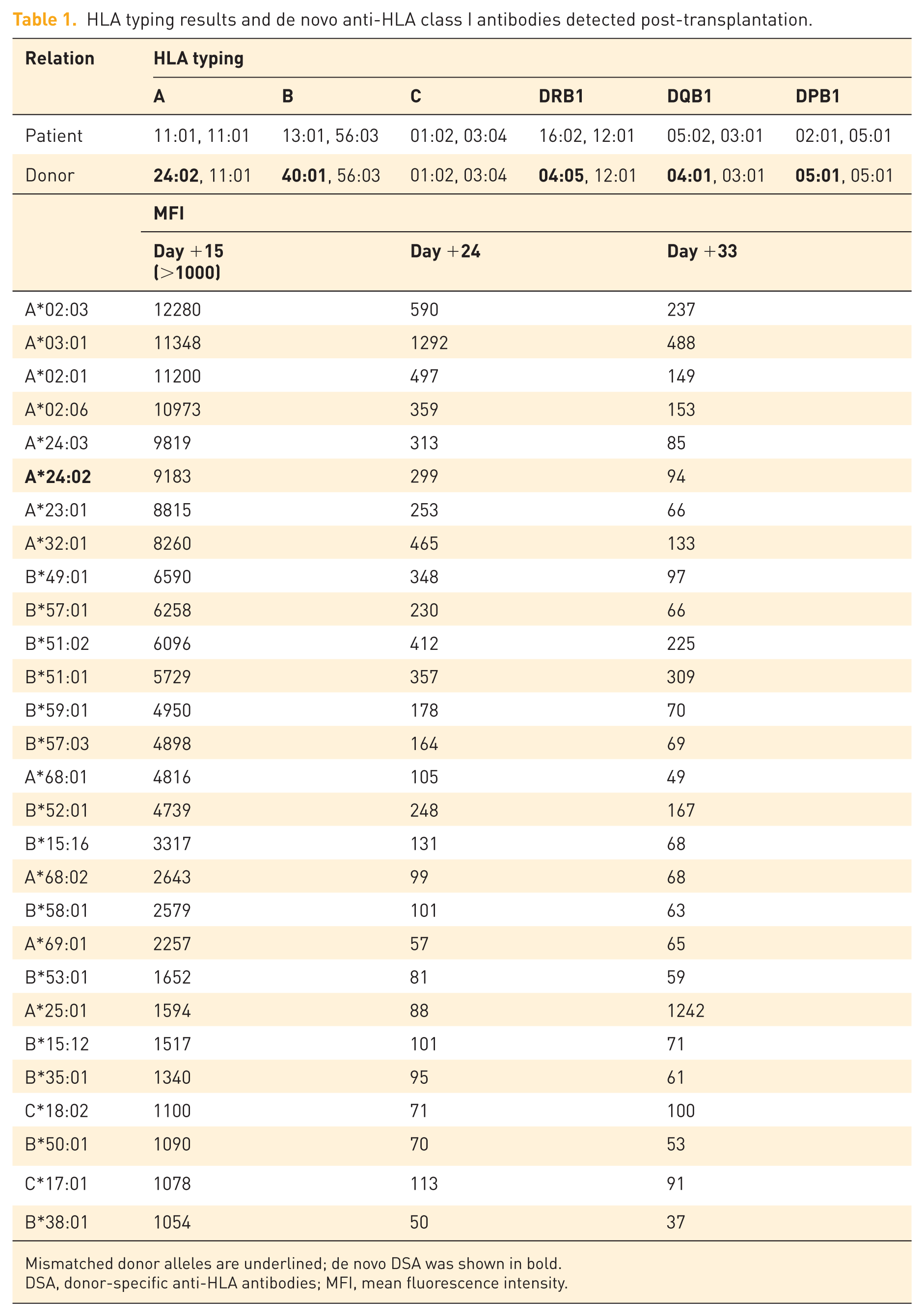
Mismatched donor alleles are underlined; de novo DSA was shown in bold.
DSA, donor-specific anti-HLA antibodies; MFI, mean fluorescence intensity.
Conditioning and transplant procedure
On September 14, 2024, the patient initiated conditioning for haplo-HSCT using a TBF + anti-thymocyte globulin (ATG)/post-transplant cyclophosphamide (PTCy) regimen. The conditioning lasted 8 days and consisted of thiotepa (5 mg/kg, days –8 to –7), intravenous busulfan (0.8 mg/kg every 6 h, days –7 to –5), and fludarabine (30 mg/m2, days –6 to –2). GVHD prophylaxis was administered using ATG (2 mg/kg, days –4 to –2), PTCy (25 mg/kg, days +3 and +4), cyclosporine (initiated intravenously at 2.5 mg/kg/day from Day –7 and subsequently adjusted according to trough levels to maintain 150–250 ng/ml), mycophenolate mofetil, and a short course of methotrexate. On day 0, peripheral blood stem cells collected from her father—including 10.19 × 108/kg nucleated cells and 8.80 × 106/kg CD34+ cells—were infused.
Development of de novo DSAs and diagnosis of primary PGF
Based on our previous data using the TBF + ATG/PTCy conditioning regimen, the median times to neutrophil and platelet engraftment in haplo-HSCT recipients were 15 and 18 days, respectively. 8 Although the TBF regimen is known to result in longer engraftment times in patients with myelofibrosis, this is generally attributable to the underlying disease itself. 9 In this patient, despite the presence of reticulin fibrosis in the bone marrow, the disease course was relatively short, the diagnosis of MDS was definitive, and no splenomegaly was observed. Nevertheless, hematopoietic recovery had not occurred by day +15 post-transplant, with a WBC count of only 0.07 × 109/L and a platelet count of 3 × 109/L, which raised significant concern. On day +15, peripheral blood chimerism testing using short tandem repeats (STR) revealed a donor cell chimerism rate of 94.58%. Simultaneously, anti-HLA antibody screening was performed, showing strong positivity for anti-HLA class I antibodies (MFI = 12,280), while class II antibodies remained negative. Notably, a DSA against HLA-A*24:02 was identified, with an MFI of 9183, whereas all HLA antibodies were negative prior to transplantation (Table 1). These findings suggested that the formation of de novo DSAs might have contributed to PGF. In response, the patient underwent three sessions of plasma exchange on days +18, +20, and +23, followed by intravenous immunoglobulin (IVIg; 1 g/kg, once) on day +24, and a single dose of rituximab (600 mg) on day +25, in an effort to reduce the DSA burden. A follow-up anti-HLA antibody test on day +24 showed a decrease in class I antibody MFI to 1292, and the A24:02 DSA level declined to 299 (Table 1). On day +33, the A24:02 DSA MFI further decreased to 94 (Table 1). Hematopoietic recovery was achieved on day +29, when the absolute neutrophil count (ANC) exceeded 0.5 × 109/L, while platelet counts remained below 20 × 109/L (Figures 1 and 2(a)).

Post-transplantation timeline of treatment and key clinical events.
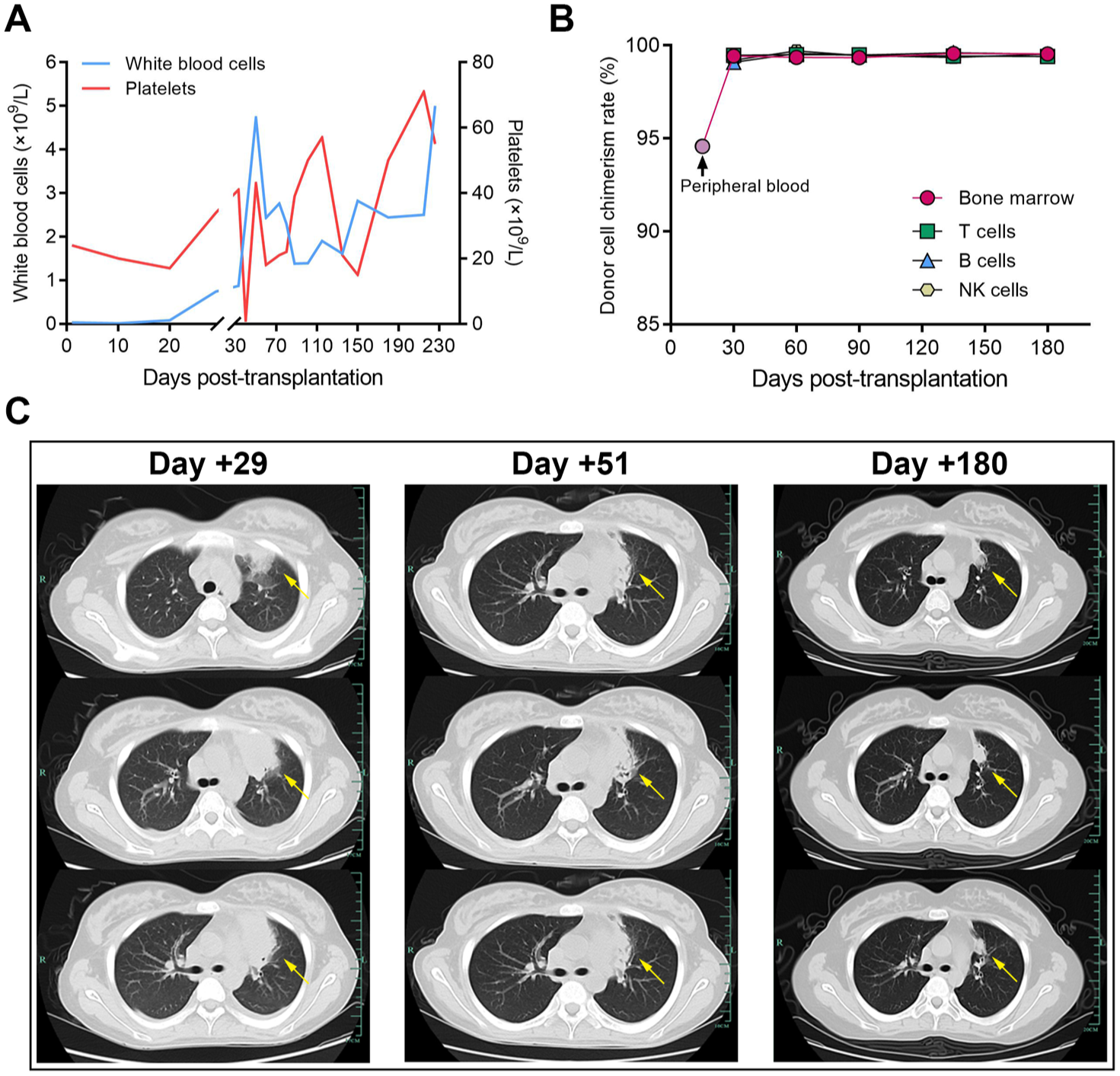
Dynamic changes in clinical parameters following transplantation. (a) Trends in white blood cell and platelet counts after transplantation. (b) Donor chimerism analysis using short tandem repeat assays post-transplantation. (c) Pulmonary infectious lesions at different stages post-transplantation.
Following neutrophil engraftment, the patient was transferred out of the transplantation ward and continued inpatient care. On day +30 post-transplantation, the patient’s ANC was 0.63 × 109/L, platelet count was 8 × 109/L—which did not meet the criteria for platelet engraftment—and hemoglobin level was 58 g/L. A bone marrow aspiration was also performed. Morphological analysis revealed a decreased number of nucleated cells, markedly hyperplastic granulopoiesis, hypoplastic erythropoiesis, and an absence of megakaryocytes. Flow cytometric immunophenotyping revealed no immunophenotypic abnormalities, and concurrently, STR analysis demonstrated a donor chimerism rate of 99.43%. Based on these findings, a diagnosis of primary PGF was established. Notably, the patient had no other common PGF-related risk factors such as an insufficient CD34+ cell dose or splenomegaly. Therefore, the early development of de novo DSAs was considered the most likely cause of PGF. In addition, the patient was diagnosed with grade 3 hemorrhagic cystitis (detailed below), which may have contributed to persistent platelet consumption and further impaired platelet recovery. Subsequent management focused on the control of transplant-related complications, the promotion of graft function recovery, and the maintenance of stable peripheral blood cell counts.
Infectious complications and immune-mediated febrile episodes
With regard to infections, from day +5 post-transplantation, the patient developed recurrent fevers, with a maximum temperature (Tmax) of 39.3°C. Despite multiple pathogen screening tests, no causative organisms were identified. Empirical treatment with imipenem/cilastatin led to temporary defervescence. However, between days +8 and +13, the patient again exhibited febrile episodes with Tmax up to 38.9°C. On day +14, persistent high-grade fever recurred (Tmax 40.2°C), and antimicrobial therapy was escalated to a combination of piperacillin-tazobactam, tigecycline, and caspofungin. Nevertheless, the fever persisted. High-sensitivity C-reactive protein peaked at 149 mg/L, while procalcitonin remained within the normal range. On day +17, metagenomic next-generation sequencing (mNGS) of peripheral blood detected Aspergillus flavus (77 reads) and Aspergillus oryzae (43 reads), and the galactomannan (GM) assay returned positive (3.42 ng/mL). Antifungal treatment was switched to voriconazole. Despite this, the patient remained febrile between days +17 and +21, with a Tmax of 40.1°C. Multidisciplinary team consultation was conducted, and in consideration of the concurrent emergence of de novo DSAs, it was hypothesized that the fever was attributable to a combination of infection and immune-mediated responses. The antimicrobial regimen was further adjusted to include ceftazidime-avibactam and daptomycin, while antifungal therapy was intensified with amphotericin B plus voriconazole. Methylprednisolone was initiated at 1 mg/kg to suppress immune activation associated with infection and de novo DSAs, with tapering based on clinical response. From day +22, the patient’s fever subsided rapidly (Tmax decreased to 37.8°C). A repeat mNGS on day +22 revealed a reduced sequence count for A. flavus (18 reads) and A. oryzae (3 reads). Throughout the febrile course, repeated cultures of blood, urine, stool, and sputum remained negative. On day +22, antibacterial therapy was switched to piperacillin-tazobactam. By day +23, the patient exhibited a marked increase in liver enzymes (ALT up to 2175 U/L) and direct bilirubin (up to 60 μmol/L). Drug-induced liver injury was suspected, most likely associated with voriconazole, which was subsequently discontinued while amphotericin B was continued as antifungal monotherapy. Meanwhile, hepatoprotective and choleretic therapies were administered as supportive measures. Thereafter, the patient’s liver enzyme and bilirubin levels gradually decreased. Between days +26 and +27, the patient experienced transient fever (Tmax 39.6°C). At that time, neutrophil counts were rising following targeted treatment for de novo DSAs, and the fever was attributed to engraftment syndrome. Symptomatic management was effective, and the patient’s temperature gradually normalized thereafter. On day +29, chest CT revealed a lesion in the upper lobe of the left lung (Figure 2(b)). The GM test remained positive. After consultation with infectious disease specialists, pulmonary aspergillosis was suspected, and antifungal therapy was switched to isavuconazole, with subsequent imaging showing gradual resolution of the pulmonary lesion (Figure 2(b)).
Management of hemorrhagic cystitis and supportive therapies for PGF
On day +33 post-transplantation, the patient presented with gross hematuria, abdominal pain, and urinary irritation symptoms, consistent with grade 3 hemorrhagic cystitis. Ultrasound revealed bilateral renal enlargement, collecting system dilation (1.5 cm right, 0.8 cm left), right-sided hydronephrosis, and bladder wall thickening with multiple echogenic clots. Abdominal CT confirmed renal swelling, bilateral hydronephrosis (predominantly right-sided), and bladder wall thickening. Urine testing detected a high BK virus load (107 copies/mL). The patient was treated with foscarnet, intensive hydration, urine alkalinization, antispasmodics, and analgesics. On day +42, intravesical instillation of cord blood-derived exosomes was performed. One month post-transplant and after discharge from the transplant unit, the patient had achieved neutrophil engraftment but remained severely thrombocytopenic (platelet nadir 1 × 109/L) and anemic (Hb 26 g/L), with no evidence of active infection or acute GVHD. This was considered due to a combination of PGF and hemorrhagic cystitis-related consumption. Supportive treatment included platelet and RBC transfusions, rhTPO, TPO-RAs, and 4 weekly infusions of cord blood-derived mesenchymal stem cells (MSCs, 1.0 × 106/kg) on days +41, +48, +55, and +62. Urinary symptoms gradually improved, and hematuria resolved. On day +58, ultrasound showed reduced right hydronephrosis (0.9 cm), no left-sided dilation, and no visible bladder clots. By day +73, the patient achieved platelet engraftment, with ANC 2.22 × 109/L and Hb 77 g/L, and was discharged in stable condition (Figure 2(a)). She was later readmitted once, at approximately +5 months post-transplant, due to CMV viremia.
Post-transplant follow-up: chronic GVHD, CMV viremia, and ongoing remission
The patient underwent regular bone marrow assessments at +2, +3, +4.5, and +6 months post-transplant, all showing complete remission and >99% donor chimerism by STR (Figure 2(c)). No acute GVHD occurred. After discharge at +2.5 months, the patient was maintained on cyclosporine (50 mg/d) and tapering methylprednisolone (12 mg/d). On day +110, moderate chronic GVHD (skin, 25%–50% BSA) was diagnosed and managed with increased cyclosporine (100 mg/d) and methylprednisolone (1 mg/kg/d), resulting in rapid improvement. Immunosuppressants were then gradually tapered. At +6 months, cutaneous cGVHD progressed (>50% BSA). Treatment was intensified with ruxolitinib (10 mg/d) and methylprednisolone (0.5 mg/kg/d) added to cyclosporine (50 mg/d), leading to marked improvement of the rash. Due to CMV viremia and thrombocytopenia, ruxolitinib was tapered and replaced with belumosudil (200 mg/d). The skin lesions gradually improved and stabilized, and immunosuppressants were progressively reduced. CMV viremia was first documented at approximately +5 months post-transplant (9.75 × 105 IU/mL), likely related to immunosuppressive therapy. Foscarnet treatment subsequently reduced the viral load to low-copy levels (~102 IU/mL). Further tapering of immunosuppressants is ongoing. At +8 months post-transplant, the patient remains in remission, without active GVHD or infection, and continues regular follow-up.
Discussion
DSAs have emerged as one of the major challenges in haplo-HSCT in recent years. The incidence of DSAs in haploidentical HSCT ranges from 10% to 21%.10–12 In a study of 345 allo-HSCT recipients, 11.3% were DSA-positive, with 31 cases involving HLA class I, 15 involving class II, and 7 involving both. 12 Female recipients have significantly higher DSA incidence (86% vs 5%) and antibody levels than males, 10 likely due to pregnancy-induced alloimmunization, especially in multiparous women with hematologic malignancies.10,13 Transfusion is another trigger, particularly with leukocyte- or platelet-rich products, which carry a higher risk of HLA sensitization than red blood cells. 14 Numerous studies have demonstrated that DSAs are closely associated with primary graft failure, PGF, GVHD, and transplant-related mortality, ultimately compromising patient survival.12,15,16 Compared with preexisting DSAs present in recipients prior to transplantation, de novo DSAs arising post-transplantation have received relatively less attention. De novo DSAs have been reported earlier and more extensively in solid organ transplantation. Numerous studies have identified de novo DSAs in recipients of kidney, heart, liver, and lung transplants. Similar to preexisting DSAs, de novo DSAs are also associated with higher rates of rejection and reduced survival. 17
Although preexisting DSAs are relatively common in HSCT recipients, reports on post-transplant de novo DSAs remain limited. A single-center retrospective study of 303 consecutive patients who underwent their first unrelated donor allo-HSCT between January 2008 and December 2017 identified 11 DSA-positive cases (3.63%), of which 10 had preexisting DSAs, and only 1 developed a de novo DSA against DPB1*04:01 (MFI 8956) on day +21 post-transplant, resulting in primary graft rejection. DSAs were the only significant predictor of primary graft failure within 28 days (HR = 2.78; 95% CI: 1.65–4.68; p = 0.0001). 18 Another retrospective study of 116 haplo-HSCT recipients without preexisting HLA antibodies found a de novo HLA antibody detection rate of 75.9% (88/116), with a median MFI of 2439 (range: 1033–20,162). Only 6.9% (8/116) had persistently positive de novo DSAs, all with MFI <2000, and none developed graft rejection or dysfunction. This study also suggests that de novo HLA antibodies—including both DSAs and non-DSAs—can be detected as early as day +15 post-transplant, and are associated with increased risk of acute GVHD (aGVHD) and reduced disease-free survial (DFS) and overall survival (OS) in patients without pre-transplant HLA antibodies. 16 Case reports have also described de novo DSA-associated graft failure or PGF in patients with chronic lymphocytic leukemia or acute myeloid leukemia following allo-HSCT.19,20 In our case, a young patient with MDS developed de novo HLA class I antibodies (MFI up to 12,280) as early as day +15 post-transplant, including a DSA against HLA-A*24:02 (MFI 9183). To the best of our knowledge, no similar cases have been reported.
The mechanism of de novo DSA formation after HSCT remains unclear. Studies in animal models and solid organ transplantation suggest that donor-reactive memory B cells play a central role. 21 De novo DSAs can emerge as early as day +15 post-haplo-HSCT, predominantly involving HLA class II antibodies. 16 In the context of HLA-mismatched haplo-HSCT, residual host T cells may recognize donor antigens and activate recipient B cells, which then differentiate into plasma cells and produce HLA antibodies. These antibodies can bind to endothelial cells and initiate signaling cascades such as FAK/Src, ERK, PI3K, and mTORC1, leading to endothelial activation and leukocyte recruitment—processes similar to vasculopathy observed in solid organ transplantation.22,23 The predominance of HLA class II antibodies may be attributed to their ability to directly recognize foreign epitopes and the high polymorphism of their encoding genes. 24 Although most de novo DSAs reported after haplo-HSCT are class II antibodies, our case presented an uncommon class I DSA, which warrants further investigation. It is also important to distinguish true donor-specific alloantibody responses from transient or polyclonal antibody elevations related to cytokine release or infection. In this patient, we identified a DSA directed against HLA-A*24:02 with a high MFI (>9000) on Day +15. The absence of systemic sepsis or cytokine storm during this period, together with the rapid decline in MFI following effective intervention, supports a true alloimmune process rather than a transient immune activation. Similar early-onset de novo DSAs have been reported in haploidentical transplant recipients by Wang et al. 16 and Ji et al., 20 emphasizing that such antibodies, even when transient, can be clinically pathogenic and associated with poor outcomes.
Current studies indicate that both preexisting and de novo DSAs are strongly associated with an increased risk of graft failure or PGF.12,15,16,18–20 Our case highlights that de novo DSAs should be considered in patients with PGF, even if they were HLA antibody-negative prior to transplantation. In such cases, DSA testing is recommended, and desensitization should be initiated promptly once de novo DSAs are detected. Although a CD34+-selected stem cell boost is a potential therapeutic strategy for PGF, in our case, the presence of high-titer de novo DSAs made such an approach unlikely to succeed. Published data suggest that additional CD34+ infusion in the setting of persistent DSAs carries a high risk of graft failure.25,26 Therefore, we prioritized desensitization and immune modulation. Reported strategies generally fall into four categories: (1) antibody removal via plasmapheresis or immunoadsorption; (2) antibody neutralization with IVIg or donor-derived HLA antigens (e.g., platelet or buffy coat transfusion); (3) inhibition of antibody production using agents such as rituximab or proteasome inhibitors, such as bortezomib; and (4) complement blockade. 25 In this case, we applied three sessions of plasmapheresis combined with IVIg and rituximab, which effectively reduced de novo DSA levels. At the same time, we actively managed other potential contributors to PGF, including infection and hemorrhagic cystitis, and administered MSCs—which are known to benefit PGF27,28—together with rhTPO and TPO receptor agonists to support platelet recovery. Timely DSA detection and comprehensive interventions ultimately led to successful stem cell engraftment in this patient.
This case provides several insights relevant to both clinical practice and future research. First, initiating DSA monitoring within the first 2 weeks after haplo-HSCT can identify clinically relevant antibodies earlier than the conventionally recommended day +28 screening time point. 16 Second, early initiation of desensitization therapy in combination with MSC infusion can reverse antibody-mediated PGF and restore hematopoiesis. Third, our observation that a class I DSA can cause severe graft dysfunction broadens the prevailing view that class II antibodies predominate, showing that class I DSAs can also be pathogenic. Collectively, these observations underscore the importance of early DSA surveillance, individualized management, and the development of standardized post-transplant DSA monitoring protocols in haploidentical HSCT.
In summary, although de novo DSAs are relatively uncommon after HSCT, their association with graft failure or PGF poses a serious threat to patient survival and warrants close attention. Early detection and timely intervention are critical to achieving successful engraftment. In parallel, prompt identification and management of transplant-related complications are also essential to ensure favorable clinical outcomes.
Limitations
This report has several limitations. First, it describes a single case, and the findings may not be generalizable to all patients undergoing haplo-HSCT. Second, mechanistic insights into how de novo DSAs contributed to PGF are limited, as functional assays were not performed. Third, although pre-transplant HLA antibody screening was negative nearly 2 months before conditioning, supportive transfusions were administered during the intervening period. Therefore, we cannot completely exclude the possibility that low-level DSAs emerged before transplantation but remained undetected at the time of conditioning. In future cases, repeat DSA testing immediately prior to transplantation, particularly when there is a delay in donor product availability or hospital admission, should be considered to detect newly formed DSAs and allow desensitization if required. Further, some therapeutic interventions, such as intravesical exosome instillation and MSC infusions, were exploratory and performed in an institutional pilot setting; their efficacy requires validation in larger prospective studies. Finally, long-term follow-up is still needed to assess the durability of engraftment, immune reconstitution, and late complications.
Conclusion
This case highlights that de novo DSAs can play a critical role in the development of primary PGF after haplo-HSCT. Timely detection and targeted immunomodulatory interventions, together with comprehensive supportive care, are essential to achieving hematopoietic recovery and favorable outcomes.
Supplemental Material
sj-jpg-1-tah-10.1177_20406207261417496 – Supplemental material for Early-onset de novo donor-specific anti-HLA antibodies may contribute to poor graft function following haploidentical transplantation for myelodysplastic syndrome: a rare case presentation
Supplemental material, sj-jpg-1-tah-10.1177_20406207261417496 for Early-onset de novo donor-specific anti-HLA antibodies may contribute to poor graft function following haploidentical transplantation for myelodysplastic syndrome: a rare case presentation by Xianbo Huang, Xianhui Wu, Shasha Wang, Yu Xu, Chen Mei, Fengwei Du, Yanling Ren, Jie Jin, Hongyan Tong and Jiejing Qian in Therapeutic Advances in Hematology
Footnotes
Acknowledgements
We are deeply grateful to the patient’s legal guardians for providing consent to share the medical records for publication. We also sincerely acknowledge the commitment and professional care of the medical staff and physicians who contributed to the management and treatment of this patient.
Declarations
Supplemental material
Supplemental material for this article is available online.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.