
Abstract
Acute leukemia (AL) is a rare yet perilous malignancy. Currently, the primary treatment for AL involves combination chemotherapy as the cornerstone of comprehensive measures, alongside hematopoietic stem cell transplantation as a radical approach. However, despite these interventions, mortality rates remain high, particularly among refractory/recurrent patients or elderly individuals with a poor prognosis. Acetylation, a form of epigenetic regulation, has emerged as a promising therapeutic avenue for treating AL. Recent studies have highlighted the potential of acetylation regulation as a novel treatment pathway. Histone deacetylase inhibitors (HDACis) play a pivotal role in modulating the differentiation and development of tumor cells through diverse pathways, simultaneously impacting the maturation and function of lymphocytes. HDACis demonstrate promise in enhancing survival rates and achieving a complete response in both acute myeloid leukemia and acute T-lymphoblastic leukemia patients. This article provides a comprehensive review of the advancements in HDACi therapy for AL, shedding light on its potential implications for clinical practice.
Plain language summary
Acute leukemia (AL) is a rare yet perilous malignancy. Presently, the primary treatments for AL encompass combination chemotherapy as the cornerstone of a comprehensive approach, and hematopoietic stem cell transplantation (HSCT) as a radical treatment. However, despite these interventions, mortality rates remain elevated, particularly among refractory/relapsing patients or older adults with a grim prognosis. Epigenetic regulation entails altering the expression of genes through pertinent genetic information without modifying the DNA sequence. Acetylation modification, as a form of epigenetic regulation, has emerged as a promising avenue for AL treatment. Recent studies have underscored the potential of acetylation regulation as a novel therapeutic approach. Histone deacetylase inhibitors (HDACis) modulate the differentiation and development of tumor cells through various mechanisms and impact the maturation and function of lymphocytes. HDACis exhibits promise in enhancing survival rates for acute leukemia, among other benefits. This article offers a comprehensive review of the advancements in HDACis therapy for AL, shedding light on its potential implications for clinical practice.
Introduction
Among blood system disorders, acute leukemia (AL) is a rare and perilous malignancy. This condition involves the malignant clonal proliferation of abnormal hematopoietic precursor cells, which accumulate within the bone marrow, consequently hampering the normal hematopoietic function of the bone marrow, and invade other vital organs, such as the liver, spleen, and lymph nodes, causing widespread damage to tissues and organs. In China, the incidence of leukemia is 3–4 cases per 100,000 individuals, and the associated mortality rate is notably high. Currently, the primary approach to treating AL involves comprehensive measures with combination chemotherapy, with hematopoietic stem cell transplantation (HSCT) serving as a radical treatment. While some patients achieve long-term survival through these interventions, the prognosis remains grim for refractory/relapsed AL patients and elderly AL patients. Fortunately, recent advancements in AL treatments offer hope for improved survival rates and reduced toxicity.
Acetylation, classified as a form of epigenetic regulation, plays a crucial role in various life processes, such as DNA damage and repair, the regulation of protein function, and cell cycle control. Such effects are achieved by modulating the acetylation level of histones. Moreover, acetylation can decrease the expression of tumor suppressor genes, thus actively participating in the progression of tumor development. In 2014, the Food and Drug Administration (FDA) approved belinostat, a deacetylase inhibitor, for treating patients with peripheral T-cell lymphoma (PTCL) who had undergone at least one prior systemic therapy, demonstrating a median response duration of 48 months. 1 Consequently, acetylation regulation holds promise as a novel therapeutic avenue for treating AL, with the potential to increase both survival and complete remission rates.
Acetylation and deacetylation
In recent years, there has been a growing focus on epigenetic events, a series of mechanisms capable of regulating gene expression patterns independently of DNA sequence mutations. 2 Epigenetic inheritance plays a vital role in governing gene expression, developmental stages, lineage commitment, and cell differentiation through various means. 3 Epigenetic mechanisms include DNA methylation, histone modification, and noncoding RNA regulation. These changes are predominantly orchestrated by enzymes such as DNA methyltransferases, histone methyltransferases, histone acetyltransferases (HATs), and histone deacetylases (HDACs). 4 Of particular interest lately is HDACs, enzymes responsible for removing acetyl groups from histones and considered pivotal regulators of gene expression. The abnormal expression of HDACs has been shown to be associated with cancer, particularly hematological malignancies. 2
Histone deacetylases
Concept and classification of HDACs
Acetylation occurs through two main mechanisms. First, irreversible N-α-acetylation involves the acetylation of the N-terminus of histones and is present in eukaryotes. The second method is reversible lysine acetylation, wherein the acetyl group from acetyl-CoA is transferred to lysine residues by acetylases.5–7 Lysine contains anchorage points that recruit specific proteins with unique domains. Histone acetylation involves the mediation of acetylation and catalysis through HATs and HDACs, maintaining a balance in histone acetylation levels. HATs promote the expression of cancer suppressor genes by altering chromatin structure, catalyzing the transfer of the acetyl group to the ε-amino group in the lysine side chain using acetyl-CoA as the common acetyl donor. This process neutralizes the positive charge of lysine residues and disrupts the electrostatic bond between DNA and histones, facilitating easier access to the local chromosomal region.5–7 Conversely, HDACs counteract the acetylation process initiated by HATs, strengthening the electrostatic force between DNA and histones and inhibiting the expression of cancer suppressor genes.8,9 Furthermore, HDACs contribute to the deacetylation of nonhistone proteins, regulating various functions, such as DNA damage repair and cell cycle regulation. 5
In humans, 18 types of HDACs are classified into four classes based on homology. Each HDAC exhibits a distinct subcellular localization model, substrate specificity, and unique enzyme activity. 10 Class I HDACs (HDAC1, 2, 3, 8) are most closely related to reduced potassium dependency 3 (RPD3) and are the most abundant HDACs located in the cell nucleus. Class II HDACs (HDAC4, 5, 6, 7, 9, and 10) can be further divided into class IIa and class IIb HDACs. Class IIa HDACs include a highly conserved C-terminal deacetylase catalytic domain homologous to that of HDAC, while class IIb HDACs possess two deacetylase domains and shuttle between the nucleus and cytoplasm with tissue specificity. Class III HDACs belong to the sirtuin family (SIRT1–7), and the activity of these HDACs is dependent on nicotinamide purine dinucleotide (NAD+). These enzymes require NAD+ as a cofactor, distinguishing them from HDACs that bind Zn2+. Some sirtuins play dual roles as tumor suppressors and oncoproteins, contributing to the ongoing debate on their function in cancer progression. Class IV HDACs are represented by HDAC11.2,11 Class I, II, and IV HDAC active sites necessitate Zn2+ as a cofactor and are inhibited by Zn2+-binding histone deacetylase inhibitors (HDACis). Class III HDACs, from a homology perspective, are markedly different from other classes. The enzymatic activities of classical HDACs (classes I, II, and IV) are not sensitive to certain inhibitors, such as vorinostat and trichostatin A (TSA). Moreover, various HDACs exhibit differential expression levels during development, growth stages, exposure to environmental stress, and hormonal stimulation.3,12
Mechanisms and application of HDACs
The different structures and classifications of HDACs endow them with a variety of capabilities. HDACs actively participate in various cellular activities, playing a crucial role in epigenetic events by influencing DNA synthesis regulation, chromatin packaging, DNA repair, and interactions with nonhistone proteins and nucleosomes.13–15 One primary domain of impact is the regulation of human blood cell production, where HDACs maintain stem cell characteristics, control the proliferation and differentiation of hematopoietic stem cells (HSCs), and guide lineage orientation. 2 HDACs modulate the expression of essential genes, promoting HSC differentiation into granulocyte–monocyte lineages, red blood cell lineages, megakaryocyte lineages, and lymphatic genealogies. Researchers widely acknowledge that normal blood production is contingent on the activity of HDACs.2,16–20 Evidence suggests that a decrease in certain HDACs can lead to blood disorders such as anemia and thrombocytopenia. Furthermore, the expression levels of HDACs may play a regulatory role in hematopoiesis. 2 An illustrative example is the continuous presence of HDAC1 throughout all stages of hematopoiesis. Maintaining low levels of HDAC1 ensures the survival and differentiation of HSCs into mature bone marrow cells, while moderate levels guide HSC development into mature red blood cells and T lymphocytes. 16
Furthermore, the upregulation of HDAC expression has been implicated in malignant tumors. Elevated levels of HDACs impede the expression of tumor suppressor genes, promoting tumor angiogenesis and migration, and ultimately resulting in the increased proliferation, invasion, and metastasis of tumor cells. For example, HDAC8 induces the deacetylation of p53, expediting the transformation of leukemia stem cells (LSCs) mediated by CBFβ-SMMHC (core binding factor β and the smooth-muscle myosin heavy chain), with the goal of sustaining self-renewal capacity and resistance to chemotherapy drugs. HDAC3 induces DNA damage and enhances drug resistance in acute myeloid leukemia (AML) cells. Consequently, inhibiting HDAC3 activity renders cells more responsive to chemotherapy drugs, reducing the likelihood of AML recurrence and progression. In summary, the mechanisms of histone acetylation are important in the context of malignant tumors. The targeted inhibition of histone acetylation may emerge as a promising avenue for tumor therapy.21–23
Histone deacetylase inhibitors
Concept and classification of HDACis
HDACis, which are recognized as potential therapeutic targets, are currently undergoing clinical trials for various diseases; they can be specific to a particular type of HDAC or can act on all HDACs. HDACis can be classified based on their chemical properties. 23 The hydroxy acid group, comprising compounds such as suberoylanilide hydroxamic acid (SAHA), TSA, LBH589 (panobinostat), and PXD101 (belinostat), can block all HDACs.24,25 The short-chain fatty acid group, widely used in clinical trials due to ease of synthesis and application in diseases, specifically targets Class I and II HDACs, with examples including valproic acid (VPA) and butyrate. However, these compounds show relative nonspecificity, with low efficacy and certain toxicity, posing challenges in achieving clinically relevant inhibitory doses. 22 The benzamide group includes MS275 (entinostat) and FK228 (romidepsin). Tetracyclic peptides can inhibit Class I, IIa, and IV HDACs. 23 Diverse structural combinations endow HDAC inhibitors with distinct chemical properties, facilitating a range of mechanisms to inhibit tumor cell proliferation, differentiation, and other processes (Table 1).
The HDACi family: classification, HDAC apecificity, mechanism, targeted genes/pathway and clinical tast.
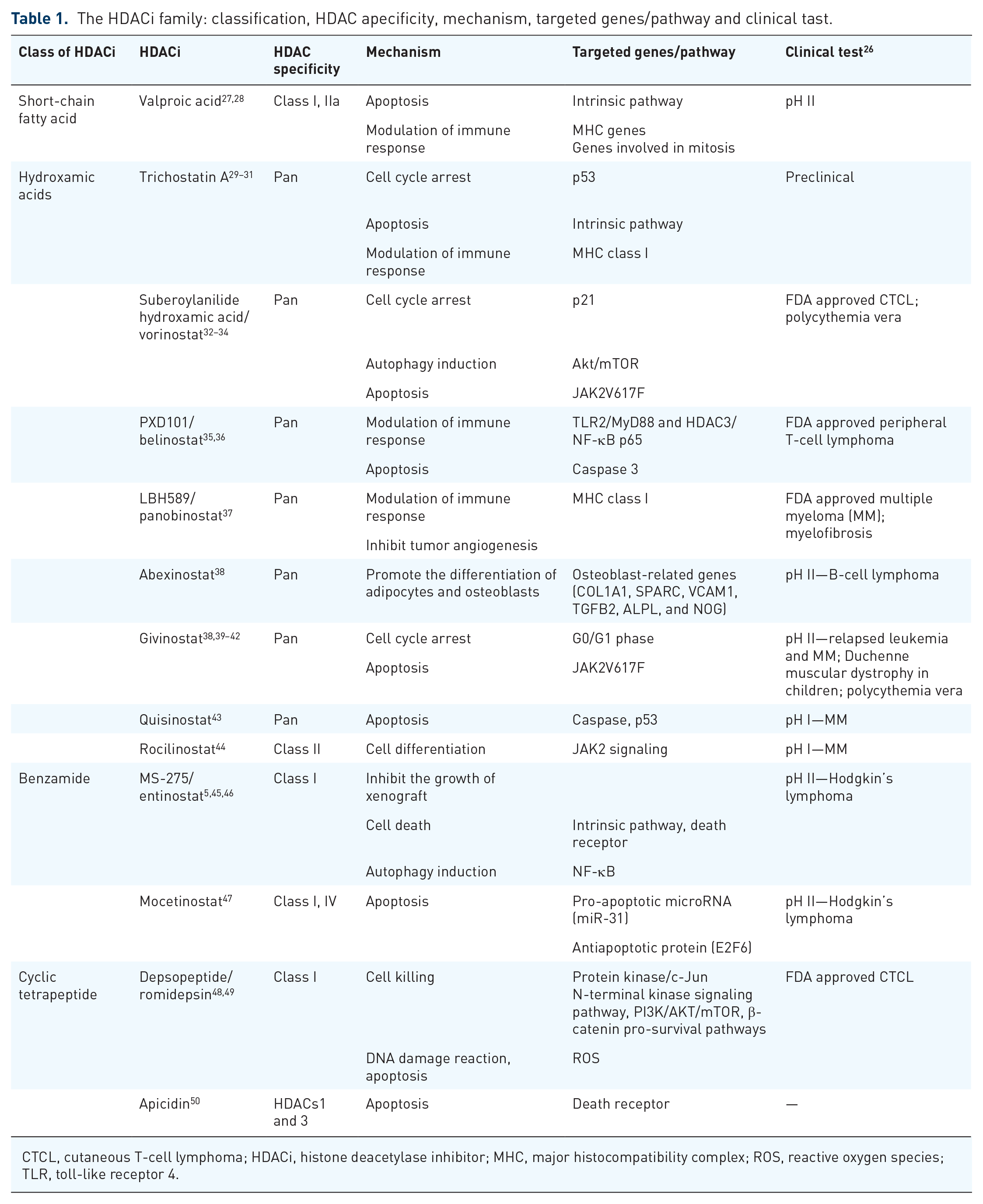
CTCL, cutaneous T-cell lymphoma; HDACi, histone deacetylase inhibitor; MHC, major histocompatibility complex; ROS, reactive oxygen species; TLR, toll-like receptor 4.
The mechanism of HDACis
Research indicates that HDACis can hinder the proliferation of various transformed cells in vitro, including lymphoma, myeloma, and leukemia cells. Moreover, they exert inhibitory effects on the progression of specific solid tumors and hematological malignancies.12,51 HDACis actively counteract the growth and development of tumor cells by inducing apoptosis through diverse mechanisms, thereby achieving therapeutic goals in cancer treatment (Figure 1).

Mechanism diagram of HDACis regulating tumor cells.
HDACis can regulate the cell cycle by diminishing cell differentiation or the release of the HDAC1 protein. 12 For instance, SAHA treats leukemia by inducing the apoptosis of leukemia cells at the G0, G1, and S stages. The hyperacetylation of histones 3 and 4 in promoter nucleosomes stimulates the genes involved in differentiation, leading to cell differentiation, apoptosis, and cycle arrest. 22 HDACis are also described as inhibitors of tumor regulation. The acetylation-mediated ubiquitylation of p53 results in cell cycle arrest and programmed death, impeding the growth and development of tumor cells. HDACis induce cell apoptosis through two mechanisms: reducing the expression of vascular endothelial growth factor receptor 2 (VEGFR-2) and influencing the VEGFR-2 signaling pathway to regulate angiogenesis. Unlike chemotherapy drugs, HDACis target both proliferating and nonproliferating cells. 52
HDACis induce apoptosis in tumor cells by activating both exogenous and endogenous apoptotic pathways. In the endogenous pathway, HDACis inactivate or inhibit antiapoptotic proteins while activating proapoptotic proteins. This process involves the release of mitochondrial intermembrane proteins due to mitochondrial destruction, ultimately leading to caspase activation. 52 Furthermore, HDACis upregulate the expression of death receptors and their ligands in transformed cells both in vivo and in vitro, while leaving their expression unchanged in normal cells. Research by Borbone et al. 53 demonstrated that the selective inhibition of HDAC1 and 2 reduces the degradation of tumor necrosis factor-related apoptosis-inducing ligand (TRAIL) proteins, inducing apoptosis in fully transformed thyroid cancer cells through the ubiquitin-dependent pathway. By downregulating the expression of antiapoptotic proteins such as caspase inhibitors and cellular FLICE-like inhibitory proteins (c-FLIP) and upregulating the expression of proapoptotic proteins such as the Bcl-2 family members Bim and Bmf, HDACis activate the death receptor pathway, leading to cleavage and activation. This connection bridges the endogenous and exogenous pathways of apoptosis. 54
HDACis have been found to induce the accumulation of reactive oxygen species (ROS) in tumor cells, leading to cell death. Studies such as that conducted by Ungerstedt et al. 55 revealed that specific HDACis, such as SAHA and MS-275 (a benzamide), promote ROS accumulation and cysteine activation in transformed cells while sparing normal cells. The distinctive mechanism underlying these effects involves increasing thioredoxin (Trx) levels in normal cells, a phenomenon not observed in transformed cells that lack Trx. In normal cells, HDACis elevate the levels of Trx, which serves as a reduced antioxidant scavenging agent for ROS. However, in transformed cells, the Trx content decreases as HDACis upregulate the expression of thioredoxin-binding protein 2 (TBP-2) and bind to Trx. 56 The impact of this interaction is significant, as observed when transformed cells were transfected with Trx small interfering RNA. In this scenario, Trx protein levels notably decreased, ROS levels increased, cell proliferation decreased, and sensitivity to SAHA-induced cell death increased. 56 This intricate interplay between HDACis, Trx, and ROS highlights a potential therapeutic avenue for selectively targeting transformed cells while preserving the viability of normal cells.
Moreover, HDACis can induce the death of tumor cells by disrupting mitosis, interfering with DNA damage repair, and altering gene expression. Although HDACis, as a method of epigenetic regulation, do not directly participate in DNA damage or gene changes, they do cause structural changes in chromatin. The exposure of DNA to damaging agents such as ultraviolet light, radiation, cytotoxic drugs, and ROS results in DNA double-strand breaks (DSBs), preventing cell division and differentiation. 52 Recent studies suggest that HDACis can directly induce DNA damage through oxidative stress and significant changes in chromatin structure rather than solely through DSBs. 54 Furthermore, HDACis disrupt the function of chaperone proteins, particularly heat shock protein 90 (Hsp90), by inhibiting HDAC6, leading to the HSP70-mediated proteasomal degradation of Hsp90 “client” oncoproteins. 54 HDACis can inhibit tumor cells and promote apoptosis through various pathways, and many of these effects are not manifested in normal cells. This selective impact offers a potential advantage for disease treatment.
Regulation of lymphocytes by HDACis
In contemporary cancer therapy, the field of lymphocyte immunity development is flourishing. Immunotherapy, including programmed cell death protein-1/programmed cell death-Ligand 1 (PD-1/PD-L1) inhibitors and chimeric antigen receptor T-cell immunotherapy (CAR-T) cell therapy, has become increasingly important for patients with advanced or refractory cancers. Histone acetylation plays a pivotal role in lymphocyte growth, development, proliferation, and differentiation, thus having significant implications for acetylation regulation in clinical antitumor therapy, particularly when combined with immune therapy (Figure 2).

Mechanism diagram of HDACis regulation of lymphocytes.
Antigen-presenting cells (APCs) are primarily immune cells that include mononuclear phagocytes, dendritic cells (DCs), B-cells, Langerhans cells, and target cells of viral infection or tumor cells, among others. LBH589, a novel panhistone deacetylase inhibitor, influences the expression of surface molecules in both immature and mature DCs, reducing DC uptake of protein antigens and inhibiting polysaccharide antigen uptake. 57 In macrophages, HDACis alter toll-like receptor 4 (TLR4)-dependent activation and function. HDACis promote the transformation of inflammatory macrophages (M1) into tolerogenic M2 cells and reduce TLR signaling.57–59 After HDACi treatment, the production of Th1 cytokines (IFN-γ and IL-2) and Th2 cytokines (IL-4) by invariant natural killer T (iNKT) cells is significantly reduced, leading to the impaired activation of iNKT cells, which is crucial for maintaining immune homeostasis.57,60,61 Second, T-cells serve as crucial mediators of tumor destruction. 62 HDACis exert their effects on the development and function of T-cells at various stages through different mechanisms. Treatment with TSA leads to histone acetylation at the IFN-γ gene locus in naïve Th0 cells and decreases the expression of CD28 in naïve CD4+ cells, significantly affecting T-cells.63,64 Multiple studies have shown that HDACs regulate initial CD4+ T-cell differentiation and cytokine release.65,66 A deficiency of HDAC1 and HDAC2 in TH0 and TH1 cells leads to the generation of CD4+ helper T-cells expressing CD8 lineage genes and a loss of the ability to inhibit runt-related transcription factor 3-core-binding factor subunit-β (Runx-CBFβ) complexes, which are crucial transcription factors for normal T-cell growth and leukemia suppression, suggesting the potential importance of combination therapy with HDACis. 67 In the context of cytotoxic T lymphocytes (CTLs), the constitutive non-TCR (T cell receptor) -regulatory pathway of CTLs targets HDAC7, controlling the expression of cytokines, cytokine receptors, and genes encoding adhesion molecules that determine CTL function. 68 Effector memory T-cells exhibit a pattern of cytokine gene acetylation under TH1 or TH2 conditions, while central memory T-cells exhibit low acetylation of cytokine genes at baseline, with increased acetylation upon stimulation, enhancing functional activity against tumors.69–71
Furthermore, Foxp3+ Tregs play a significant role in various immune processes, including transplant rejection, autoimmunity, and allergies. 72 The presence of HDACs or HDACi complex components in Tregs may contribute to the regulation of Foxp3 acetylation, increasing the stability of Tregs. 73 Among the various classes of HDAC enzymes, Class II and Class III HDACs have proven to be particularly valuable in this context. 74 The immunosuppressive nature of AML is closely linked to Tregs, particularly those expressing tumor necrosis factor receptor 2 (TNFR2). A study demonstrated a reduction in TNFR2(+) Treg levels in both peripheral blood and bone marrow after exposure to panobinostat and azacitidine for TNFR2(+) Tregs in AML patients. 75 Zhao et al. 76 found that in patients with immune thrombocytopenia purpura (ITP), low-dose chidamide increased the number of Treg cells in a peripheral blood mononuclear cell culture system, inhibiting the proliferation of effector T-cells, suggesting that low-dose chidamide has a significant therapeutic effect on ITP. Zuo et al. 77 reported that the median fluorescence intensity of Treg cells was significantly reduced after chidamide combination treatment with chidamide and chemotherapy for T-cell non-Hodgkin lymphoma. Notably, Tregs can inhibit graft-versus-host disease (GVHD). Specifically, targeting HDAC6 and HDAC9 can enhance Treg function, leading to the clearance of residual tumor cells without intensifying the GVHD effect. This targeted inhibition is significant for managing the delicate balance between GVHD and graft-versus-leukemia, improving graft acceptance and ultimately increasing the transplantation success rate.78,79
HDACis for AL
HDACis for AML
AML is the most common form of AL in adults and typically develops at an older age. The median age of onset is approximately 70 years, and the survival rate for elderly patients is generally poor.80,81 The primary treatments for AML include standard chemotherapy and allogeneic HSCT. However, the effectiveness of these treatments is limited, and resistance to chemotherapy drugs often occurs, leading to a high recurrence rate. 82
In recent years, the role of HDACis in AML treatment has become clearer. AML is sustained by LSCs, with the core-binding factor (CBF) complex being a crucial transcription factor targeted in AML. The inv(16) (p13q22) chromosome inversion leads to the formation of the CBFβ-SMMHC fusion protein, enhancing mutations in the cytokine signaling pathway during leukemia transformation. 83 Comprising the DNA-bound RUNX protein and non-DNA-bound CBFβ protein, the CBF complex is vital for hematopoietic function. CBFβ fuses with the SMMHC protein, forming the fusion protein CBFβ-SMMHC (CM), which contains the RUNX1 binding interface of CBFβ and the helix-helix-rod region of SMMHC. Additionally, RUNX1 plays a crucial role in HSC production and serves as a key transcription factor for hematopoietic cells. HDAC8 binds to CM protein, increasing p53 deacetylation, leading to p53 inactivation, reducing the impact of p53 on LSCs, and promoting LSC proliferation. 80 Therefore, inhibiting HDAC8 effectively restores p53 acetylation and activity. inv(16) generates the CBFβ-SMMHC fusion protein, which is prevalent in the acute myeloid leukemia M4 subtype with eosinophilic cells (M4Eo). 84 Notably, HDAC8 inhibition induces apoptosis in AML cells with inv(16), significantly decreases AML cell proliferation, and eliminates leukemic initiation ability in mouse- and patient-derived LSCs while preserving normal HSCs. 85 Another study revealed that HDAC1 and CM can form complexes that colocalize with RUNX1 and CBFβ-SMMHC on the promoters of known fusion protein target genes. 86 In a mouse model, entinostat, an HDAC1 inhibitor, was found to reduce leukemia burden in vivo, inducing leukemia cell differentiation and apoptosis. Therefore, HDAC has emerged as a promising target for effective AML treatment.
VPA, a short-chain fatty acid and HDACi, induces the differentiation of AML progenitor cells in vitro by acting as a proliferation inhibitor and apoptosis inducer in AML cells. The combination of all-trans retinoic acid (ATRA) enhances the effects of VPA. 87 VPA/ATRA therapy shows promise for transiently controlling disease progression in AML evolving from myelodysplastic disease (MDS). However, a clinical trial in 26 low-risk AML patients using VPA + ATRA yielded no complete responses (CRs), with only mild improvement observed in a neonatal AML patient. 88 Further exploration is required for primary AML treatment with VPA + ATRA. Chidamide selectively induces apoptosis in LSC-like cells and primary AML CD34+ cells in a concentration- and time-dependent manner. 89 Mechanistically, chidamide triggers LSC death by activating ROS, disrupting the mitochondrial membrane potential, modulating BCL2 family proteins, and activating caspase-3, resulting in poly ADP-ribose polymerase (PARP) degradation. Additionally, chidamide activates CD40 and regulates the downstream signaling pathways JNK and NF-κB, suggesting that chidamide is a potential novel LSC-targeted AML therapy. In another study, Ma et al. 90 explored the impact of a novel HDAC inhibitor, I13, on inducing differentiation in M3 and M5 subtype AML cells, t(8; 21) translocated M2 subtype AML cells, and leukemia stem-like cells. I13 disrupts the cell cycle, induces differentiation, and significantly inhibits the proliferation and colony formation of AML cells. Consequently, I13 has emerged as a potential alternative compound capable of overcoming the blocking of AML differentiation.
While the preclinical results of HDACis show promise, the monotherapy effect of HDACis on AML is modest. The combination of HDACis with multiple drugs represents a novel approach to AML treatment. One study assessed the therapeutic efficacy and feasibility of combining VPA with low-dose cytarabine (Ara-C) for treating elderly and frail AML patients. 91 Among the 31 patients, 8 achieved sustained CRs, and 3 achieved hematological improvement, resulting in an overall response rate (ORR) of 35% with relatively low toxicity. This treatment option can be considered for patients who are unable to undergo standard induction therapy. The resistance of AML to conventional therapy significantly reduces cure rates, and Khateb et al. 92 reported that the increased expression and abundance of the ubiquitin ligase RNF5 contribute to AML development and survival. The inhibition of RNF5 can reduce the growth of AML cells in culture and in vivo, enhance the sensitivity of AML cells to HDACis, and prolong the survival of mice. One study reported the effects of a novel HDACi, LBH589, combined with adriamycin on AML cells. 93 Panobinostat exhibited extensive anti-AML activity. Additionally, panobinostat combined with adriamycin-induced AML cell death by increasing mitochondrial outer membrane permeability and releasing mitochondrial cytochrome c, leading to Caspase-dependent apoptosis. This mechanism is likely to trigger cell death through a mechanism that induces DSBs. Therefore, the combination of panobinostat with doxorubicin may prove to be an effective treatment for acute myeloid leukemia. Vorinostat is not effective as a single agent in the treatment of AML but has some efficacy in combination therapy. 94 A phase II clinical trial evaluated the safety and efficacy of the HDACi vorinostat in combination with bortezomib and Ara-C in patients with AML or MDS. The study enrolled 75 previously untreated AML or high-risk MDS patients aged 15–65 years with appropriate organ function and no CBF abnormalities. The overall survival (OS) rate was 82 weeks, and the ORR was 85%, with a 76% CR rate and 9% CR with incomplete platelet recovery. 95 Because HDACis can enhance the efficacy of gemtuzumab ozogamicin in vitro, a phase I/II trial explored the efficacy of gemtuzumab ozogamicin combined with vorinostat and azacitidine in the treatment of refractory/relapsed elderly patients with AML. 96 Among the 43 patients, 10 achieved a CR, and 8 achieved a CR with incomplete platelet recovery, resulting in an ORR of 41.9%. Furthermore, Akada et al. 32 have demonstrated that vorinostat possesses therapeutic potential for the treatment of polycythemia vera (PV) and other myeloproliferative neoplasms linked to the JAK2V617F mutation. Vorinostat markedly inhibited the proliferation and induced apoptosis in cells expressing JAK2V617F. Concurrently, it potently suppressed the growth of mouse and human PV hematopoietic progenitor cells harboring the JAK2V617F mutation. Notably, givinostat, an orally administered HDACi, gained its initial approval on March 21, 2024, in the United States, for the management of Duchenne muscular dystrophy in patients aged 6 years and above. 39 Givinostat is a potent inducer of apoptosis and death of multiple myeloma cells, 38 at the same time, is also studies on the treatment of relapsed leukemia. 40 Amaru Calzada et al. 41 propose that a combined regimen of givinostat and hydroxyurea holds promise as a novel strategy for the treatment of Jak2(V617F)-driven myeloproliferative neoplasms. This development indicates that extensive investigation into HDACis like vorinostat and givinostat may open up novel avenues for the management of a broader spectrum of hematological disorders. In Europe, decitabine is also approved for older AML patients with >20% untreated cells, but the treatment has been less effective, with CR rates of 9%–18% in randomized phase III clinical trials. Another phase I trial evaluated the efficacy of continuous or simultaneous administration of vorinostat plus decitabine every 28 days in patients with relapsed/refractory AML. 97 The continuous regimen involves the continuous administration of decitabine followed by vorinostat, while in the concurrent regimen, patients are given both decitabine and vorinostat. The ORR was 23%, with a reliable safety profile. Among the two regimens, the response rate was greater for the concurrent regimen.
HDACis for ALL
ALL is another type of AL. The overall incidence of ALL is 3.85 per 100,000 people, with 60% of patients being under 19 years old. 5 The primary treatment for ALL consists of multiagent chemotherapy, but its efficacy remains limited for patients with relapsed/refractory ALL. Allogeneic HSCT is considered an ideal therapy for ALL; however, recurrence posttransplantation significantly diminishes its effectiveness. Considering the challenges in ALL treatment, there is interest in exploring the potential use of HDACis for ALL patients. Current investigations into HDACis are largely inspired by their application in PTCL. Romidepsin, which was approved by the US FDA in November 2009 for cutaneous T-cell lymphoma (CTCL) treatment, showed promising results in a pivotal open-label phase II study for relapsed or refractory PTCL. 98 In this study, patients were treated for 28 days, resulting in an ORR of 25%, with CR/complete remission unconfirmed accounting for 15%. The median duration of overall response was 17 months. Notably, controllable adverse events (AEs) were observed, and serious AEs such as thrombocytopenia, neutropenia, and anemia were rare. Consequently, romidepsin has received approval from the US FDA for treating relapsed/refractory PTCL. 99
In July 2014, belinostat gained FDA approval for treating patients with relapsed or refractory PTCL, while vorinostat obtained FDA approval for managing relapsed/refractory CTCL in 2006.1,100–102 A multicenter, open-label, pivotal phase II study demonstrated that chidamide, used for relapsed or refractory PTCL, achieved an ORR of 28%, with a median progression-free survival (PFS) and OS of 2.1 and 21.4 months, respectively. In December 2014, the China Food and Drug Administration approved chidamide for the treatment of ALL. 103 In vitro, HDACis have been combined with various drugs, including topoisomerase inhibitors, bortezomib, and cytotoxic chemotherapy agents. Numerous clinical studies are underway to explore the efficacy of these combinations in treating T-cell lymphoma. 100 Given the promising therapeutic outcomes of HDACis in PTCL and the extensive research conducted, it is reasonable to consider that HDACis could prove beneficial for ALL patients, especially those with a poor prognosis.
HDACis and acute T-lymphoblastic leukemia
Acute T-lymphoblastic leukemia (T-ALL) is considered an aggressive subtype of ALL. 104 Some studies have shown promise in combining HDAC inhibitors with chemotherapy or HSCT to improve survival outcomes in patients with T-ALL. Zhou et al. 105 conducted an open, single-arm, multicenter clinical trial to investigate the efficacy and safety of chidamide in treating adult early T-cell precursor acute lymphoblastic leukemia (ETP-ALL). The study included 24 patients with ETP-ALL, comprising 4 females and 20 males, with a median age of 22 years (14–22 years). The photodynamic therapy (PDT)-ETP-ALL regimen plus 10 mg/day of chidamide was employed from induction therapy to consolidation therapy. The results revealed an 87% CR rate after induction therapy. Six out of 24 (25%) ETP-ALL patients underwent allo-HSCT, and the 2-year event-free survival (EFS) rate was 83%. Guan et al. 82 conducted a comparative study evaluating the efficacy of chemotherapy alone versus chidamide combined with chemotherapy in treating relapsed refractory T-ALL patients. The findings demonstrated that the combined treatment group exhibited a superior CR rate, ORR, and PFS compared to the chemotherapy-only group. However, there was no significant difference in OS between the two groups. In a prospective study, Li et al. 106 confirmed that chidamide, when combined with chemotherapy, demonstrated good clinical efficacy and safety in treating T-ALL in children. The OS and EFS were 94.1% and 95.2%, respectively, in the chidamide treatment group. A small-sample study conducted at Guangdong Provincial People’s Hospital indicated that maintenance chemotherapy with chidamide (10 mg twice weekly) for five patients with NOTCH1- and RAS/PTEN-mutated adult T-cell lymphoblastic lymphoma (LBL) resulted in no relapses at the last follow-up (122.0 months). 107
Patients with T-ALL who undergo allo-HSCT still face a substantial risk of recurrence posttransplantation, which remains the primary cause of death in T-ALL patients following this procedure. The potential role of maintenance therapy with targeted drugs posttransplantation in mitigating the risk of recurrence warrants comprehensive clinical investigation. In one study, the efficacy of a combination of 0.5 mg/kg Chidamide administered once every 4 days for the treatment of T-ALL in children was explored. 106 Maintenance therapy commenced 4–8 weeks after transplantation and extended for up to 2 years posttransplantation. Following maintenance treatment, the ORR and EFS were reported to be 94.1% and 95.2%, respectively. Additional studies have investigated the efficacy and safety of low-dose decitabine in preventing relapse after allo-HSCT for ALL in adults. The results indicated that among 12 T-ALL/LBL patients, the 2-year relapse rate, OS, and disease-free survival rates were 8.3%, 90%, and 81.5%, respectively, with no recurrence observed in seven T-ALL patients. 108
HDACis and B-ALL
Acute B-lymphoblastic leukemia (B-ALL) patients are treated with chemotherapy. However, in high-risk patients, relapse or metastasis to extramedullary tissues or organs occurs within a year, and the 5-year survival rate is generally only approximately 35%. The sensitivity of NALM-6 human B-ALL cells to spiruchostatin B (SP-B), a histone acetylase inhibitor, has been observed. The expression of P21-related mRNAs in these cells was significantly greater than that in other types of leukemia cells. SP-B inhibits histone acetylation and activates the caspase cascade, leading to NALM-6 cell apoptosis. Notably, prior to the induction of apoptosis by SP-B, the expression of P21-related mRNA in NALM-6 cells increases, facilitating SP-B recognition and the induction of leukemia cell apoptosis. 109 The HDACi LAQ824 has a similar effect on NALM-6 leukemia cells. 110 A study by Agirre et al. 111 confirmed the impact of LBH589 on mouse models of T-ALL and B-ALL. Compared with those treated with vincristine and dexamethasone, mice treated with LBH589 showed increased levels of H3 and H4 acetylation and survived longer. Notably, the combination of LBH589 with vincristine and dexamethasone significantly enhanced the treatment efficacy. Similarly, Mehrpouri et al. discovered that LBH589 prolongs the G1 phase of cells by increasing C-MYC-mediated cyclin-dependent kinase inhibitors, thereby reducing the viability of B-ALL cells. Additionally, they found that bortezomib synergistically enhances the antitumor effects of LBH589 by inhibiting the NF-κB pathway, suggesting that LBH589 is a potential therapeutic agent for B-ALL. 112 In the treatment of children with ALL, the metabolism of the commonly used antifolate methotrexate (MTX) is dependent on folylpolyglutamate synthetase (FPGS). Chromatin remodeling can alter the sensitive sites of FPGS upstream of exons, thus modifying FPGS expression. Research indicates that the HDACis sodium butyrate and SAHA can increase FPGS mRNA expression by 2–5 times without enhancing the cytotoxicity of MTX. Therefore, the combination of HDACis and MTX has the potential to improve therapeutic efficacy in clinical ALL patients. 113
In contrast to generalized HDACi research, Stubbs et al. 114 aimed to identify the HDACi to which B-ALL is most sensitive. They assessed the sensitivity of B-ALL cells to various subtypes of HDACis and found that HDAC6 inhibitors, which selectively inhibit HDAC6 in the high-dose range, are effective against B-ALL cells. Both in vitro and in vivo, the selective inhibition of HDAC1 and HDAC2 significantly impeded the growth of B-ALL cells, with little effect on the growth of other hematopoietic malignant tumors. Consequently, the development of a drug that selectively inhibits HDAC1 and HDAC2 holds promise as a targeted therapy for precision treatment of B-ALL. In 2019, Yang et al. 115 evaluated the preclinical efficacy of purinostat mesylate, a novel class I and IIb HDAC inhibitor, for treating breakpoint cluster region (BCR)-abelson proto-oncogene (ABL) induced Ph+ B-ALL. Purinostat mesylate was found to downregulate the expression of BCR-ABL and c-MYC, inducing apoptosis in primary Ph+ B-ALL cells in Ph+ leukemia cell lines and relapsed patients. Furthermore, purinostat mesylate effectively impeded the progression of Ph+ B-ALL, significantly extending the survival of BL-2 secondary transplantation models with clinical symptoms of Ph+ B-ALL, BCR-ABL(T315I)-induced primary B-ALL mouse models, and BCR-ABL-specific tyrosine kinase inhibitor-induced recurrent Ph+ B-ALL patients. These findings suggest that purinostat mesylate may serve as an effective treatment for B-ALL patients and patients with relapsed/refractory disease.
Conclusion
The conventional approach for treating AL primarily relies on combination chemotherapy, which has limitations in drug selection and is often associated with adverse reactions. The efficacy of this treatment, especially for recurrent/refractory patients, is modest at best. At present, the forefront of research on blood system tumor treatment is centered on epigenetic modifications, with a particular emphasis on rapid advancements in HDACis. HDACis operate at both the molecular and cellular levels, regulating cell differentiation and apoptosis, inducing cell cycle arrest, and modulating the function of immune cells such as T-cells and APCs. This multilevel intervention in tumor cell formation and differentiation, along with the induction of apoptosis, suggests that HDACis are a promising avenue for AL treatment. Some HDACis have already received approval for hematological malignancies, while others are undergoing investigation as monotherapies or adjunct agents for their anti-AL effects. Moreover, the combination of HDACis with chemotherapy, targeted therapy drugs, immunotherapy drugs, or CAR-T cell therapy has demonstrated enhanced effectiveness in treating AL. This has led to the emergence of new antitumor inhibitors, such as the dual matrix metallopeptidase 2 (MMP2)/HDAC-8 inhibitor, an antitumor substance metabolizer developed for AML and ALL by Halder et al. 116 These innovations are expected to provide improved options for treating AL. Additionally, there is substantial room for further exploration into how HDACis influence gene replication, transcription, translation, and chromatin remodeling. This ongoing research may unveil more therapeutic strategies involving HDACis, offering additional directions for treating hematologic tumors. The dynamic landscape of HDACis in the field holds great promise for advancing the treatment of AL.
The use of HDACis in the treatment of AL is still in its early stages, and caution is warranted, especially considering the current trend toward personalized medicine for AL. Based on current research findings, monotherapy with HDACis has not shown satisfactory results in AL patients. Further investigation is essential to understand the precise mechanisms by which different types of HDACis act on various subtypes of AL tumors. Rigorous and extensive in vivo studies are necessary to establish the safety and reliability of HDACis before they can be used in more effective clinical trials. Moreover, some approved HDACis lack selectivity, leading to off-target effects and undesirable side effects. The development of certain HDACis in clinical trials has been hindered by poor pharmacokinetic properties. 51 Efforts to address these limitations have been made, as demonstrated by Fan et al., 117 who explored prodrug strategies. Prodrugs are inactive or partially active drugs that become active parent drugs in the body through enzymatic or chemical reactions. Prodrug strategies aim to overcome the shortcomings of the physical and chemical properties of HDACis, enhancing their absorption, distribution, metabolism, excretion, and other characteristics. However, challenges arise due to the biotransformation of precursor drugs and the degradation of parent drugs and ligands/active groups, which are influenced by various internal and external factors. Additionally, when combining HDACis with other drugs, it is crucial to clarify the drug toxicity and compatibility issues associated with these combinations. Further exploration of the interdependent mechanisms between tumor epigenetics and tumor immunology or metabolism is needed. Anticipating increased investment in future research, there is a growing focus on developing HDACis as potential treatments for AL patients and expanding their application to a broader spectrum of patients with hematological malignancies.