
Abstract
Chimeric antigen receptor T-cell (CAR-T) therapy, which has demonstrated notable efficacy against B-cell malignancies and is approved by the US Food and Drug Administration for clinical use in this context, represents a significant milestone in cancer immunotherapy. However, the efficacy of CAR-T therapy for the treatment of acute myeloid leukemia (AML) is poor. The challenges associated with the application of CAR-T therapy for the clinical treatment of AML include, but are not limited to, nonspecific distribution of AML therapeutic targets, difficulties in the production of CAR-T cells, AML blast cell heterogeneity, the immunosuppressive microenvironment in AML, and treatment-related adverse events. In this review, we summarize the recent findings regarding various therapeutic targets for AML (CD33, CD123, CLL1, CD7, etc.) and the results of the latest clinical studies on these targets. Thereafter, we also discuss the challenges related to CAR-T therapy for AML and some promising strategies for overcoming these challenges, including novel approaches such as gene editing and advances in CAR design.
Plain language summary
Acute myeloid leukemia (AML) remains a clinical challenge despite the advent of chimeric receptor T-cell (CAR-T) therapy, as there are obstacles hindering the application of CAR-T cells in AML. In this review, we discuss the results of current relevant clinical trials, existing treatment strategies for AML and recent advances in preclinical research to provide insight for overcoming the inefficacy of CAR-T therapy for AML.
Introduction
Acute myeloid leukemia (AML) is characterized by an increase in the number of immature myeloid progenitor cells (blasts) in the bone marrow or peripheral blood and is associated with chromosomal abnormalities such as t(15;17), t(8;21), and t(16;16) or genetic mutations. 1 Most of the AML blasts and leukemia stem cells (LSC) highly express myeloid cell surface antigens, such as CD33, CD123, and CLL1 (C-type lectin-like molecule-1). The prevalence of AML increases with age, and it is the most common leukemia in adults. 2 Due to a relatively high prevalence and low 5-year survival rate, the treatment of AML has long been a research hotspot. Conventional therapeutic approaches, like chemotherapy and hematopoietic stem cell (HSC) transplantation (HSCT), have been used since the 1970s; with the further elucidation of the pathogenesis of AML, the option of targeted therapy, including the use of US Food and Drug Administration-approved agents such as midostaurin [an FMS-like tyrosine kinase 3 (FLT3) inhibitor], venetoclax (a B-cell lymphoma 2 inhibitor), and gemtuzumab ozogamicin (an antibody–drug conjugate), has also become available. One of the most notable advances with regard to immunotherapy strategies is the development of chimeric antigen receptor (CAR) T-cell (CAR-T) therapy. T cells collected from donors or the patient are artificially engineered to express CARs that can induce host immune cells to recognize and eliminate tumor cells after ex vivo expansion and injection into the patient’s body. However, although CAR-T therapy has had considerable success in treating B-cell lymphocyte leukemia and lymphoma,3,4 the evidence for CAR-T therapy being effective and safe for AML is less convincing, mainly due to the absence of ideal treatment targets, various factors related to the AML microenvironment, and phenomena such as CAR-T exhaustion.
Nonetheless, despite the factors currently limiting the applications of CAR-T therapy for AML, it remains a promising area of research, and numerous clinical trials on various related strategies have been completed or are in progress (Table 1). In this article, we will review the state-of-the-art of CAR-T therapy for AML, with a focus on therapeutic targets and novel approaches that may help overcome the limitations associated with its use for this purpose.
Currently recruiting clinical trials on CAR-T therapy for AML.

Data source: https://clinicaltrials.gov/ (accessed 11 November 2023).
AML, acute myeloid leukemia; BPDCN, blastic plasmacytoid dendritic cell neoplasm; CAR-T, chimeric antigen receptor T cell; CLL1, C-type lectin-like molecule-1; FLT3, FMS-like tyrosine kinase 3; ILT3, Ig-like transcript 3; MDS, myelodysplastic syndrome; MM, multiple myeloma; NHL, non-Hodgkin lymphoma; NKG2D-L, natural killer group 2 member D ligand; NKL, natural killer cell lymphoma; T-ALL, T-cell acute lymphoblastic leukemia; TIM-3, T-cell immunoglobulin and mucin domain-containing protein 3; T-LBL, T-cell lymphoblastic lymphoma.
Therapeutic targets and corresponding clinical trials
Any therapeutic target for AML should ideally meet all of the following criteria to ensure good treatment outcomes: (1) high expression on the surface of AML blasts; (2) minimal expression in normal tissue; (3) stable expression to prevent antigen escape; and (4) high expression on LSCs to avoid recurrence.5–7 Potential therapeutic targets in AML and the corresponding clinical trials are listed in Table 2.
Current clinical trials and results.
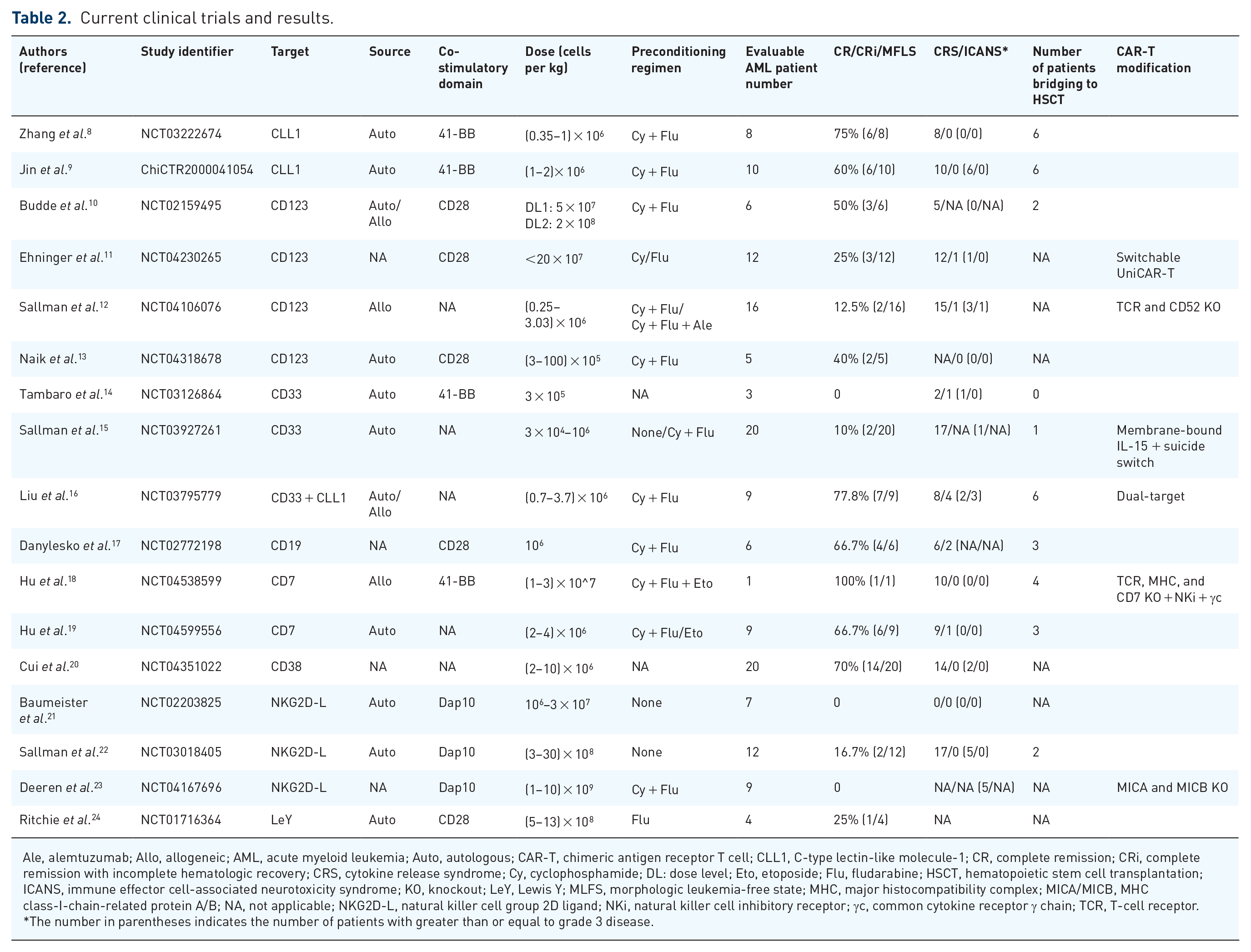
Ale, alemtuzumab; Allo, allogeneic; AML, acute myeloid leukemia; Auto, autologous; CAR-T, chimeric antigen receptor T cell; CLL1, C-type lectin-like molecule-1; CR, complete remission; CRi, complete remission with incomplete hematologic recovery; CRS, cytokine release syndrome; Cy, cyclophosphamide; DL: dose level; Eto, etoposide; Flu, fludarabine; HSCT, hematopoietic stem cell transplantation; ICANS, immune effector cell-associated neurotoxicity syndrome; KO, knockout; LeY, Lewis Y; MLFS, morphologic leukemia-free state; MHC, major histocompatibility complex; MICA/MICB, MHC class-I-chain-related protein A/B; NA, not applicable; NKG2D-L, natural killer cell group 2D ligand; NKi, natural killer cell inhibitory receptor; γc, common cytokine receptor γ chain; TCR, T-cell receptor.
The number in parentheses indicates the number of patients with greater than or equal to grade 3 disease.
CD33
CD33 (Siglec-3) is a transmembrane glycoprotein belonging to the sialic acid binding Ig-like lectin family. It is reported to be highly expressed on both AML blasts and LSCs but is not expressed on HSCs.25–27 Although its high expression on blasts and LSCs makes it an attractive target, it is also expressed on normal myeloid progenitor cells and downstream myeloid cells. 28
The initial results of the first clinical trial on CAR-T33 (NCT01864902) were published in 2015. 29 This trial involved a 41-year-old male patient diagnosed with relapse/refractory AML (r/rAML) who received CAR-T33 cells derived from his peripheral blood. The blast ratio decreased remarkably to <6% after infusion; however, it gradually increased and rose to approximately 70% at 9 weeks, and the patient died due to disease progression at 13 weeks post-infusion. In another clinical study (NCT03126864) by Tambaro et al., 14 three patients screened for r/rAML received autologous CAR-T33 treatment. Unfortunately, none of them responded well to the therapy. Cytokine release syndrome (CRS) and immune effector cell-associated neurotoxicity syndrome (ICANS) were observed in two and one patients, respectively, and all patients eventually died due to disease progression. Notably, 10 patients were initially enrolled in this study, but seven of them could not undergo infusion due to apheresis because of AML progression in two cases, CAR-T cell manufacturing failure in three cases, and disease-related death before infusion in one case. These cases highlight challenges such as rapid AML progression and T-cell dysfunction in patients with AML; thus, a shorter CAR-T cell manufacturing process and the quality of T cells are critical for CAR-T therapy to be used effectively for treating patients with AML.
CD123
CD123, the alpha subunit of the interleukin-3 (IL-3) receptor, functionally binds to IL-3 under physiological conditions and is primarily expressed on myeloid cells and over-expressed on AML blasts. In addition, it can be found on the surface of LSCs, 25 and there is also evidence to suggest that it is expressed on a small number of HSCs 30 as well as normal cells, including hematopoietic progenitor cells, dendritic cells, monocytes, and endothelial cells. 31 Although the expression pattern of CD123 makes it a possible AML therapy target, the potential of CAR-T123 toxicity due to expression in normal tissues cannot be ignored.
The first-in-human CAR-T123 therapy clinical trial (NCT02159495) published its initial results in 2017. 10 Six patients with r/rAML were included and received different doses of engineered cells. Three patients achieved complete remission (CR), and two of them subsequently bridged to a second allo-HSCT. Toxicity during treatment was well controlled, with five patients experiencing grade 1–2 CRS, one patient developing adenoviral pneumonia, and one patient experiencing a grade 3 rash. Treatment-related cytopenia and dose-limiting toxicity were not observed in any patient. In recent years, universal CAR (UCAR)-T has garnered attention due to advantages such as short preparation time, lack of restriction on patient T-cell quality, and off-the-shelf availability. It is a type of CART prepared using T cells from healthy volunteers rather than those from patients. However, in the two clinical trials (NCT04106076 and NCT04230265) on UCAR-T cells targeting CD123, the CR/complete remissions with incomplete hematologic recovery (CRi) rates were only 12.5% (2/16) and 25% (3/12).11,12
CD19
CD19 is specifically expressed in B cells (both normal and malignant), making it an ideal target for CAR-T therapy, especially for B-cell malignancies, including B-cell acute lymphoblastic leukemia (B-ALL) and lymphoma, and anti-CD19 CAR-T therapy has yielded good outcomes.3,4 In addition, AML cells may abnormally express CD19 in patients with t(8;21)(q22;q22) AML 32 ; therefore, CD19 CAR-T therapy could be effective in the treatment of t(8;21) r/rAML.
In a phase II clinical trial (NCT02772198) that included six patients with t(8;21) r/rAML, all patients experienced grade 1–3 CRS after infusion, and two patients developed ICANS. On day 28 of evaluation, four (66.7%) patients achieved CR, while the remaining two showed no response to CAR-T19 therapy. Unfortunately, three of the four patients with CR experienced disease recurrence and died due to disease progression or sepsis. 17 Although CD19 CAR-T therapy has shown some efficacy in a few trials, further large-scale clinical trials are needed to confirm its effectiveness and safety.
CD7
CD7, a member of the immunoglobulin superfamily, is expressed on AML blasts – in one study, flow cytometry analysis detected CD7 on blasts in approximately 10–40% of patients with AML using by – and has been recognized as a marker of aggressive disease, poor treatment response, and poor prognosis. 33 Its aberrant expression on AML blasts suggests that CD7 can be a potential target for CAR-T therapy.
Hu et al. 18 conducted a clinical trial (NCT04538599) on a modified CD7 CAR-T product named RD13-01 that was designed to be CD7 negative (to avoid fratricide) after genetic depletion, T-cell receptor (TCR) negative to avoid graft-versus-host disease (GvHD), and human leukocyte antigen class II-negative to avoid allo-rejection. RD13-01 was administered to 12 patients, including 1 AML patient with a pre-treatment CD7-positive blast percentage of 75%. After infusion, grade 1 CRS occurred, but no sign of ICANS was observed. The patient achieved CRi on day 28, eventually bridged to allo-HSCT, and remained in CR for over 10 months in the follow-up period. 18 Zhang et al. have recently presented preliminary data from their universal CD7 CAR-T clinical trial (NCT04599556) on nine AML patients. No dose-limiting toxicity was observed during treatment, and although one patient developed grade 1–2 ICANS and all patients experienced grade 1–2 CRS and grade 3–4 hematological toxicity, all of these conditions resolved spontaneously or after administrating tocilizumab/dexamethasone. In terms of efficacy, six (66.7%) patients achieved CR/CRi after infusion, with five remaining in remission for a median follow-up time of 5.4 months. 19
CD38
CD38, a type 2 transmembrane glycoprotein, is predominantly expressed in plasma cells, activated lymphocytes, natural killer (NK) cells, and myeloid precursor cells but is not expressed on HSCs. 34 In one study, it was reported to be expressed on AML blasts in all samples from 37 patients with AML, indicating its potential as a target for CAR-T therapy. 35
The latest results from a CAR-T38 clinical trial on 20 patients with r/rAML (NCT04351022) showed that 14 (70%) patients achieved CR/CRi after therapy, including 10 (50%) patients who further achieved minimal residual disease (MRD) negativity. In terms of safety, all adverse events were controllable (14 cases of CRS, including 2 cases of grade 3 CRS). Reversible grade 3–4 hematologic toxicity was observed in most patients, but no signs of ICANS were detected. 20
CLL1
CLL1 is a type II transmembrane glycoprotein that is selectively expressed on 78–92% of AML primary cells and most LSCs, but not on normal HSCs and only on a few lymphoid progenitor cells. 36
Zhang et al. 8 conducted a phase I/II multicenter study involving eight children with r/rAML. Six of them achieved CR/CRi/morphologic leukemia-free state (MLFS), of which four were MRD negative and two were MRD positive. Four children were still alive and continued to exhibit CR at the last follow-up. During treatment, all patients experienced grade 1–2 CRS, but ICANS was not observed. In another trial (ChiCTR2000041054) conducted by Jin et al. 9 involving 10 adult r/rAML patients, seven patients achieved CR/Cri, and six of them were alive at the end of the follow-up, with a median follow-up period of 206 days, ranging from 80 to 424 days. After CAR-T infusion, 60% of patients experienced high-grade CRS, but no patients experienced CAR-T therapy-related encephalopathy syndrome. Severe pancytopenia occurred in all patients, and two patients died of severe infection due to chronic neutropenia. Miao et al. 37 recently reported that an 18-year-old male patient with r/rAML who received haploidentical donor-derived anti-CLL1 CAR-T therapy achieved CR, bridged to allo-HSCT from the same donor and achieved MRD negativity, and subsequently maintained this state for 6 months. Liu et al. 16 designed dual-target (CLL1 and CD33) CAR-T cells, which yielded a 77.8% (7/9) CR rate in an early phase I clinical trial (NCT03795779).
Overall, the available data suggest that CLL1 is a specific and potentially effective target for AML treatment.
NKG2D-L
Natural killer group 2 member D ligand (NKG2D-L) is almost absent in normal healthy tissues, but it is upregulated in infected cells and tumors, including in AML, and mediates the activation and functioning of immune cells by interacting with NKG2D and itself during infection and in cancer.38,39
However, the results of clinical trials on NKG2D-CAR-T therapy for AML treatment have been disappointing. In the first-in-human trial (NCT02203825) on NKG2D-CAR-T, treatment-related adverse events such as CRS, ICANS, and death did not occur, but none of the 12 patients with AML or multiple myeloma responded to the therapy. Due to the poor efficacy of the response, it was suggested that increasing the dose or performing multiple infusions may increase the expansion of the cells. 21 The subsequent clinical trial (NCT03018405, product name: CYAD-01) with higher doses and multiple CAR-T infusions had similar results, with only 2 out of 12 (16.7%) patients with AML achieving CR/CRi. 22 The second-generation CAR-T product CYAD-02, which resists fratricide due to the downregulation of NKG2D-L expression by short hairpin RNA, 40 was used in a phase I clinical trial (NCT04167696) involving lymphocyte depletion preconditioning. Of the 11 enrolled patients with r/rAML or myelodysplastic syndrome (MDS), two patients with MDS achieved marrow CR, but no patients with AML achieved CR/CRi. 23
Other targets
In addition to the seven targets mentioned above, numerous other therapeutic AML targets have been effective in preclinical trials. These include Lewis Y (LeY), 41 FLT3, 42 CD44 isoform variant 6, 43 folate receptor β, 44 Siglec-6, 45 CD70, 46 CD13, 47 and leukocyte immunoglobulin-like receptor B4 (LILRB4). 48 The CR rate in the clinical trial on LeY (NCT01716364) was 25% (1/4), but the remission was transient. 24 Studies targeting FLT3 (NCT05023707, NCT05445011, NCT05432401, and NCT05017883), Siglec-6 (NCT05488132), CD70 (NCT04662294), and LILRB4 (NCT04803929) are currently recruiting patients, and their results are yet to be reported.
Reasons for CAR-T treatment failure and strategies for overcoming various limitations
AML blast heterogeneity and multi-target therapy
AML is well known for its heterogeneity, which might partially explain the high recurrence and the ability of AML cells to escape immune detection; thus, it is not surprising that single-target strategies are not successful in eliminating AML blasts. 49 One approach to solving this problem is using OR-gated CAR-T therapy, which is a generalized multi-target therapy involving several different forms of CAR-T cells, such as cells co-expressing two different CARs or CARs with two different forms of single-chain variable fragment (scFv), or tandem CARs, to increase CAR-T-cell sensitivity by broadening their recognition spectrum. 50 Xie et al. 51 recently designed a CD123 and CLL1 dual-targeted CAR-T therapy (123CL-CAR-T) and reported that it exhibited robust in vivo and in vitro antitumor activity against AML cells expressing either of the two targets. Dual-target CAR-T cells are also the subject of a clinical trial (NCT03795779). 16 According to the latest report, nine patients with r/rAML received an infusion of CAR-T cells targeting both CLL1 and CD33 and demonstrated a 77.8% (7/9) MRD negativity rate within 1 month of infusion. The adverse events included eight cases of CRS (two cases of grade 3 CRS) and four cases of ICANS (three cases of grade 3 ICANS). Another multi-target strategy is to combine CAR-T cells with switch molecules. Nixdorf et al. 52 established a Fab-based adapter CAR (AdCAR)-T platform for AML that can switch the CAR-T targets using adaptor molecules of different antigen-binding regions. In their preclinical study, AdCAR-T cells were able to target three well-known AML targets (CD33, CD123, and CLL1) and exhibited more flexible and enhanced killing of different AML cells and heterogeneous AML blasts in vitro, indicating considerable potential for clinical application.
Antigen downregulation
Downregulation of tumor targets is an important reason for CAR-T resistance and disease recurrence 53 ; thus, upregulating these targets is a possible strategy for directly protecting against AML antigen loss. Multiple drugs have been reported to upregulate the expression of tumor-associated antigens (TAAs) in AML cells. Yoshida et al. 54 reported that all-trans retinoic acid could upregulate the expression of CD38. Azacitidine has been observed to increase the expression of CD123 and CD70,55,56 and valproic acid and gilteritinib can upregulate the expression of NKG2D-L on AML blasts.57,58
Multi-target strategies are another approach to mitigate tumor escape due to antigen downregulation. In addition to the OR-gated CAR-T therapy mentioned above, IF-BETTER-gated CAR-T therapy is another choice. This involves using a fully functional CAR and a chimeric costimulatory receptor (CCR) targeting another TAA. In tumors that simultaneously express CCR and CAR targets, CCR can compensate for the loss of CAR-T-cell activation when the CAR target level is low. Compared with OR-gated CAR-T therapy, IF-BETTER gating reduces off-tumor toxicity and provides a wider selection of CCR targets, as the presence of CCR targets alone does not activate CAR-T cells. 50 Haubner et al. 59 designed IF-BETTER-gated CAR-T cells targeting adhesion G protein-coupled receptor E2 as the CAR and CLL1 as the CCR. These CAR-T cells were shown to achieve better elimination of AML cells even when CAR target levels were low and were found to outperform both single-target CAR-T and OR-gated CAR-T cells.
Improving manufacturing processes
There are two main types of CAR-T cells: patient-derived (autologous) and donor-derived (allogeneic) cells (Figure 1). Autologous cells are associated with some limitations: (1) the manufacturing process is time-consuming, and patients may fail to undergo apheresis due to rapid disease progression; (2) T cells collected from patients may be dysfunctional; and (3) the cost is high. 60 Allogeneic CAR-T cells address these issues to some extent, but they are associated with additional problems, such as allogeneic rejection and GvHD. 60

Comparison between autologous and allogeneic CAR-T cells.
UCAR-T cells refer to allogeneic, modified CAR-T cells derived from healthy donors that have the advantages of a shorter manufacturing process, less restrictive indications, and relatively lower costs. 61 Generally, to reduce the risk of GvHD/allo-rejection, UCAR-T cells that are TCR- and/or MHC-I/II-negative are generated using gene editing technology. 62 UCAR-T cells have been used in a clinical trial (NCT04106076) wherein 16 r/rAML patients received UCART123v1.2 cells negative for TCR and CD52; two of the patients achieved CR/MLFS. 12
Most of the allogenic CAR-T products mentioned above are based on αβ-dominated T cells, which require gene editing to deplete alloreactive TCRs to avoid allogeneic rejection and GvHD. However, gene editing cannot eliminate TCRs and may lead to problems such as chromosomal translocation. 63 γδT cells are also allogeneic CAR-T candidates that do not cause GvHD because they contain non-alloreactive TCRs, due to which their TCR gene does not need to be knocked out. 64 Two clinical trials on CAR γδT cells for AML treatment have been conducted (NCT04796441 and NCT05388305) and have shown that γδT cells engineered to express specific CARs have higher anti-AML activity than their CAR-negative counterparts. 65
In addition to selecting T cells derived from allogeneic sources, researchers have also tried to shorten the manufacturing process. PRGN-3006 is an optimized CAR-T33 product obtained using an overnight manufacturing process that skips the step of ex vivo expansion 66 ; it is currently being evaluated in a phase I clinical trial (NCT03927261). A total of 24 patients (20 with r/rAML, 3 with MDS, and 1 with chronic myelomonocytic leukemia) were enrolled; the total CR/CRi rate was 10% (2/20) for AML patients, with one patient achieving CR with MRD negativity after allo-HSCT and maintaining this status for 18 months. 15
Minimizing on-target, off-tumor toxicity
The nonspecific distribution of AML antigens prevents CAR-T cells from differentiating between normal cells and AML blasts, resulting in ‘on-target, off-tumor toxicity’. Capillary leak syndrome, due to the expression of CD123 on endothelial cells, is an example of a severe form of on-target, off-tumor toxicity during CAR-T123 treatment. 67
The classical CAR is composed of a typical extracellular antigen-binding domain, a transmembrane domain, and an intracellular signal transduction domain (CD3ζ). To enhance the antitumor effects of CAR-T cells and prolong cell survival, one or two T-cell costimulatory molecules, such as CD28 and 41BB, are artificially incorporated into the intracellular domains of CARs to obtain what are known as second- or third-generation CARs, respectively. 68 Both fourth- and fifth-generation CARs are based on second-generation CARs. CAR-T cells based on fourth-generation CARs, also known as T cells redirected for universal cytokine killing (TRUCK), can also induce the accumulation of T cells and innate immune cells to mediate antitumor activity due to engineered transcription factors that induce the expression of cytokines (e.g. IL-12). Fifth-generation CARs contain IL-2Rβ in their intracellular domain, which can induce Janus kinase–signal transducer and activator of transcription 3/5 (JAK-STAT3/5) pathway activation and subsequently increase CAR-T-cell proliferation and antitumor activity. 69 To circumvent on-target, off-tumor toxicity and achieve good clinical antitumor effects, many novel CAR designs have been developed (Figure 2). Some of these are discussed here.
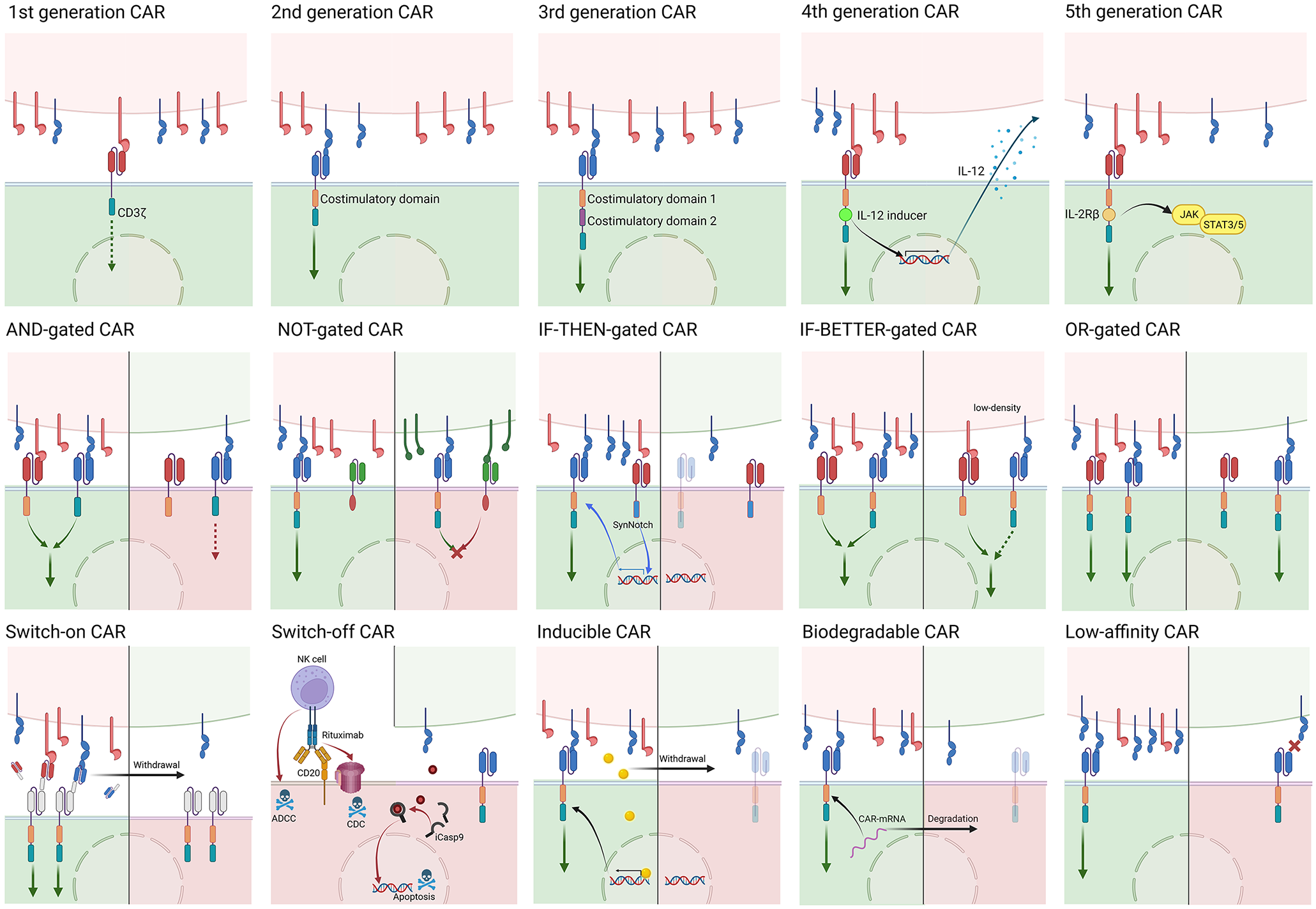
The top row shows the five generations of CARs: All five CARs share the same extracellular domain [a heavy (VH) and a light variable chain (VL) connected by a linker] and transmembrane domain. The intracellular signaling domain of first-generation CARs consists of only CD3ζ, while a costimulatory domain (CD28/41BB/OX40) is incorporated in second-generation CARs; moreover, third-generation CARs have two costimulatory domains in the intracellular region. Both fourth- and fifth-generation CARs are based on second-generation CARs: fourth-generation CARs, also known as TRUCK CARs, induce pro-inflammatory cytokine (e.g. IL-12) secretion thanks to an engineered immunostimulatory transgene. Fifth-generation CARs contain IL-2Rβ in its intracellular domain, which can induce JAK-STAT3/5 pathway activation. The middle and bottom rows show innovative CAR designs. In each panel, the upper portion represents normal cells (green) or AML blasts (red), and the lower portion represents CAR-T cells in different states (green: activated; red: inactivated or dead).
Switchable CARs
Off-switch strategies involving the use of molecules or drugs to hamper the function of CAR-T cells in a timely manner have been devised to reduce off-tumor toxicity and other side effects. The suicide gene strategy mainly involves the transduction of apoptotic genes such as inducible caspase9 and RQR8 (an artificially generated gene for T cell combining target epitopes from both CD34 and CD20 antigens) into cellular therapeutic products, enabling them to be ‘switched off’ (via apoptosis, etc.) in a timely manner when an exogenous dimerization inducer or monoclonal antibody (such as rituximab) is administered.70,71 For instance, Sommer et al. 70 inserted two rituximab mimotopes into the extracellular domain of a CAR such that they can bind to rituximab and rapidly mediate the depletion of CAR-T cells.
On-switch strategies that utilize special molecules bridging CARs and TAAs can also control off-tumor toxicity. UniCAR-T only recognizes a short peptide motif called the targeting module (TM), which can bind to tumor antigens via a scFv. Thus, off-tumor toxicity can be controlled by suspending the administration of TM, which has a short half-life and is cleared from the body rapidly. Cartellieri et al. 72 administered UniCAR-T cells targeting CD123/CD33 to AML models and observed TM-dependent tumor lysis and CAR-T-cell proliferation both in vitro and in vivo. The results of a phase I clinical trial (NCT04230265) on UniCAR-T cells targeting CD123 were recently reported – consistent with preclinical evidence, adverse events, including 12 cases of CRS (including 1 case of grade 3 CRS), 1 case of grade 2 neurotoxicity, and 1 case of DLT, could be controlled and reversed by withdrawing TM. In terms of efficacy, three patients achieved CR/CRi, including one with MRD negativity. 11 Compared to off-switches, on-switch effectors are gradually excreted from the body, leading to temporary CAR-T dysfunction until the next administration of the exogenous switch, thus avoiding one of drawbacks of off-switches, that is, induction of irreversible cell apoptosis rather than temporary blocking of CAR-T function.
Logic-gated CARs
Developed based on the concept of Boolean logic, logic-gated CARs usually consist of two separate CAR structures that recognize different antigens. AND-gated CARs comprise one first-generation CAR structure and a CCR (without CD3ζ). An effective signal that activates CAR-T cells is transduced only when two different tumor antigens are simultaneously targeted. 73 Preclinical evidence indicates that AND-gated CAR-T cells exhibited higher AML selectivity and less toxicity toward HSCs compared to single-target CAR-T cells.74,75 Like AND-gated CARs, IF-THEN-gated CARs also require the simultaneous expression of two different targets, but with sequential activation through a constitutive synthetic Notch (SynNotch) receptor and a conditionally expressed CAR that is regulated by the former. When the SynNotch receptor recognizes a tumor target, transient CAR expression is induced by the transcription factor cleaved from the SynNotch receptor. Moreover, as the CAR used in the IF-THEN gate strategy is not constitutively expressed, it can also prevent CAR-T cells from exhaustion. 76 Finally, NOT-gated CARs comprise a fully functional CAR and an inhibitory CAR (iCAR) targeting an antigen with high expression on normal tissues and limited expression on tumors. The iCAR blocks the function of CAR-T cells by transducing inhibitory signals when normal tissue is recognized. 73 Richards et al. 77 designed NOT-gated anti-CD93 CAR-T cells with an iCAR targeting CD19. CD93 is expressed on both AML and endothelial cells, and theoretically, anti-CD93 CAR-T cells should exert off-tumor toxic effects on endothelial cells. However, with NOT-gated CAR-T cells, researchers could protect endothelial cells from toxicity in vitro by engineering them to express CD19 while maintaining CAR-T toxicity against CD93+ AML cells. In addition, they also identified some iCAR target candidates, including platelet endothelial cell adhesion molecule 1 and tyrosine kinase with immunoglobulin-like and epidermal growth factor-like domains 1, two canonical endothelial markers, using single-cell RNA sequencing of AML and endothelial cells.
Other novel CAR-T therapy designs
Other strategies include electroporating CAR33-RNA into T cells to create a ‘bio-degradable’ CAR whose levels gradually decrease as the RNA is degraded, 78 substituting a single residue in the CAR binding region to lower the affinity of the CAR (low-affinity CAR-T), 79 and regulating the expression of CARs by administering exogenous molecules (inducible CAR-T). 80 Biodegradable CAR was assessed in a clinical trial (NCT02623582), but the trial was terminated in 2017 due to a lack of evidence of efficacy. 81
Gene editing
A novel gene editing solution was developed by Kim et al. 28 by depleting the expression of CD33 on HSCs using CRISPR/Cas9. They reported that not only did CD33 knockout not disrupt the normal functioning of HSCs and myeloid cells, but that it also increased the resistance of these cells to CAR-T33 in CD33-KO HSC-engrafted rhesus macaques and CD33-KO HSC-engrafted mice. Gene editing strategies allow the development of novel HSCT-based approaches for AML treatment, as they can be used to generate HSCs with higher CAR-T tolerance prior to CAR-T infusion, enabling the CAR-T cells to eliminate AML blasts with a minimized risk of off-tumor toxicity against HSCs. Currently, a clinical trial on gene editing for AML (NCT05945849) is underway, and it will be interesting to see whether this novel form of CAR-T therapy bridged to HSCT can outperform conventional HSCT bridged to CAR-T therapy.
Counteracting the immunosuppressive microenvironment
Previous studies have shown that the tumor microenvironments, including the AML microenvironment, suppress T-cell activity and proliferation.82,83 The involved mechanisms include cellular interaction between T cells and immunosuppressive cells [myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), regulatory T-cells (Tregs), and mesenchymal stromal cells], anti-inflammatory factors, and metabolic alteration.84–93 In addition, some preclinical evidence suggests that the immunosuppressive factors mentioned above also negatively impact CAR-T therapy results by dampening CAR-T cytotoxicity and proliferation.94–96 A phase I trial (NCT03355859) reported that TAM infiltration was negatively associated with the remission status of B-cell non-Hodgkin lymphoma patients during CD19 CAR-T treatment. 97 Another clinical trial (NCT02735291) highlighted that a high Treg level indicated poor prognosis for patients with refractory/relapsed B-ALL receiving CD19 CAR-T therapy. 98 Whether the AML microenvironment negatively impacts CAR-T treatment outcomes in clinical trials needs further investigation, but the preclinical evidence and clinical evidence from studies on acute lymphoblastic leukemia and lymphoma indicate that the AML microenvironment could provide clues for improving CAR-T therapy effectiveness. Strategies targeting immunosuppressive cells are likely to be feasible. CD33 has been reported to be expressed on the surface of AML blasts and MDSCs, making it a promising target that can have MDSC clearance ability and direct toxic effects against CD33+ AML blasts. 99 Eckard et al. 100 observed that a CD33/CD3 bispecific T-cell engager (AMV564) not only depleted MDSCs but also stimulated T-cell proliferation and enhanced the efficacy of CD8+ T cells, which might improve the effectiveness of CAR-T therapy. Moreover, increased AML cell toxicity and prolonged survival of tumor-bearing mice were observed when IL-2 diphtheria toxin, which can deplete Tregs, was administered to the mice prior to AML-specific cytotoxic T cells. 101
Immune checkpoints play crucial roles in immune tolerance under physiological conditions as well as in various malignancies, including AML.102,103 In addition to cytotoxic T-lymphocyte-associated protein 4, programmed cell death protein 1, and T-cell immunoglobulin and mucin domain-containing protein 3, immune checkpoint proteins such as CD70, B7-H3, and LILRB4 are reported to be expressed at high levels in AML blasts but low levels in normal tissues.48,104,105 Thus, designing a CAR that directly targets immune checkpoints seems to be an attractive strategy. Anti-CD70/B7-H3/LILRB4 CAR-T cells have exhibited robust anti-AML activity and low toxicity in normal tissues in preclinical trials,48,106,107 and clinical trials on the treatment of AML using CD70 CAR-T cells (NCT04662294) and LILRB4 CAR-T cells (NCT04803929) are currently recruiting. Immune checkpoint blockade (ICB) is another option that has been explored, but significant beneficial effects of the combination of ICB and CAR-T therapy were not observed in preclinical trials. 108 The use of CAR-T cells and immune checkpoint depletion has also been attempted. 109 In addition, Ma et al. 110 applied CLL1 CAR-T cells with PD-1 knockdown to two AML patients who relapsed after allo-HSCT. They both reached MRD-negative CR or Cri after treatment.
Conclusion
CAR-T therapy is being increasingly investigated as an immunological strategy for AML treatment based on its considerable success in treating B-cell malignancies. In this article, we have briefly summarized AML therapeutic targets and the results of relevant clinical trials and then discussed the two major limitations of CAR-T treatment in AML, that is, poor efficacy and safety issues. The poor efficacy of CAR-T therapy in treating AML is attributed to various characteristics of AML blasts, the AML microenvironment, and CAR-T cells themselves. We also focused on on-target, off-tumor toxicity, another major cause of concern related to the treatment of AML using CAR-T therapy due to the nonspecific distribution of AML targets. Compensatory strategies such as improving and optimizing CAR design have been shown to increase the efficacy and safety of CAR-T therapy in preclinical trials, and some modified CARs have already entered clinical testing. In addition, novel tools such as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR-associated protein 9 have further facilitated the application of CAR-T therapy for AML. In conclusion, CAR-T therapy is a promising approach for AML treatment, but further research is needed to overcome its limitations.