
Abstract
Chimeric antigen receptor T-cell (CAR-T) therapy has been approved for relapsed/refractory B-cell lymphomas and greatly improves disease outcomes. The impressive success has inspired the application of this approach to other types of tumors. The relapsed/refractory T-cell malignancies are characteristic of high heterogeneity and poor prognoses. The efficacy of current treatments for this group of diseases is limited. CAR-T therapy is a promising solution to ameliorate the current therapeutic situation. One of the major challenges is that normal T-cells typically share mutual antigens with malignant cells, which causes fratricide and serious T-cell aplasia. Moreover, T-cells collected for CAR transduction could be contaminated by malignant T-cells. The selection of suitable target antigens is of vital importance to mitigate fratricide and T-cell aplasia. Using nanobody-derived or naturally selected CAR-T is the latest method to overcome fratricide. Allogeneic CAR-T products and CAR-NK-cells are expected to avoid tumor contamination. Herein, we review the advances in promising target antigens, the current results of CAR-T therapy clinical trials in T-cell malignancies, the obstacles of CAR-T therapy in T-cell malignancies, and the solutions to these issues.
Keywords
Introduction
Prognosis and current treatment for T-cell malignancies
T-cell malignancies are a highly heterogeneous group of diseases with poor prognoses that stem from T-cell precursors as well as mature T-cells and can be classified into T-cell leukemia and peripheral T-cell lymphomas (PTCLs). 1 T-cell acute lymphoblastic leukemia (T-ALL) accounts for approximately 15% to 25% of ALL cases, and PTCLs account for approximately 15% of non-Hodgkin’s lymphoma (NHL) cases.2,3 Overall, traditional chemotherapy remains the first-line treatment for T-cell malignancies.
A study showed that after induction therapy, T-ALL had a similar rate of complete remission (CR) (94% versus 93%) and 5-year overall survival (OS) (48% versus 42%) as B-cell acute lymphoblastic leukemia (B-ALL). But the rate of relapse at 5 years after CR was as high as 42%, and only 8 of 123 relapsed patients survived after 5 years of follow-up. 4 This showed that although the response rate was nearly 50%, patients with T-ALL were prone to relapse. The salvage chemotherapy regimen for relapsed T-ALL varies and includes nelarabine, anthracycline-based regimens, methotrexate-based regimens, etc. In a study that enrolled 118 patients with R/R T-ALL who received nelarabine salvage therapy, the OS at 1 year of the patients was 37%, and the median survival was 8 months. 5
Generally, the patients who underwent relapse had dismal outcomes with no more than 10% of them surviving at 5 years. 6 Allogeneic hematopoietic stem cell transplantation is a potential method for curing leukemias; however, the 3-year OS after HSCT was only about 30%. 7
Another study demonstrated that PTCL was also prone to relapse. The median time from primary therapy to relapse or progression was 6.7 months. Moreover, the OS and progression-free survival after relapse were only 5.5 months and 3.1 months, respectively. 8 Current treatments for T-cell lymphomas other than traditional cytotoxic chemotherapy include histone deacetylase inhibitors (HDACi), such as chidamide, romidepsin, and belinostat; monoclonal antibodies, such as brentuximab vedotin (BV) and mogamulizumab; and other agents, such as pralatrexate and nelarabine.9–13 Using the anti-CD30 monoclonal antibody BV plus CHP (cyclophosphamide + doxorubicin + prednisone) as the primary therapy for CD30 + PTCLs has been demonstrated to be effective and superior to CHOP (cyclophosphamide + doxorubicin + vincristine + prednisone) chemotherapy. 14 However, a study found that no improvement in FFS1 and OS1 was observed after the administration of romidepsin and pralatrexate in patients with R/R diseases. 15 A similar result was reported in another study in which adding romidepsin to the CHOPE (cyclophosphamide + doxorubicin + vincristine + prednisone + etoposide) regimen in the first-line treatment of PTCLs did not exhibit benefits. 16 The OR and CR of the anti-CCR4 antibody mogamulizumab for relapsed PTCLs were only 35% and 14%, respectively, in a phase II study. 17 HSCT is the only probable method to cure this disease, but approximately 1/4 to 1/3 of patients are not eligible for HSCT. Moreover, patients who undergo HSCT are confronted with graft-versus-host disease that could lower the quality of life and even be lethal. 18 A study showed that the 5-year OS rates of salvage therapy through autologous and allogeneic HSCT were 32% and 52%, respectively, whereas the OS rate for patients who did not accept transplants was only 10%. 15 The dismal outcomes of R/R T-cell malignancies call for more efficient therapies to ameliorate the current therapeutic situation.
Chimeric antigen receptors
Chimeric antigen receptors are fusion receptors that comprise the antigen-recognition domain and T-cell signaling domain. 19 The antigen-recognition domain (the extracellular part) is the antibody-derived single-chain variable fragment (scFv) that can target the tumor antigen and thus navigate T-cells to the tumor site. The intracellular part is composed of a T-cell receptor CD3ζ subunit in the first generation of chimeric antigen receptors (CARs). In the second and later generations of CARs, a costimulatory domain is introduced into the cells to enhance their activation and function20–22 [Figure 1(a)–(c)]. Manufacturing sufficient numbers of functional chimeric antigen receptor T-cell (CAR-T) cells is the first step of CAR-T therapy. T-cells are collected by leukapheresis and then genetically transduced with a gene that contains CAR sequences by retrovirus or lentivirus. 23 After CAR-T-cell proliferation and lymphodepletion chemotherapy, eligible patients receive CAR-T-cell infusion. Currently, anti-CD19 CAR-T-cells have been successfully administered and improved the prognoses of patients with R/R B-cell malignancies, such as CD19+ diffuse large B-cell lymphoma (DLBCL), mantle cell lymphoma (MCL), and B-ALL.24–27 Anti-BCMA (B-cell maturation antigen) CAR-T products were approved by Food and Drug Administration (FDA) for treating R/R multiple myeloma. In the clinical trials, they demonstrated impressive efficacy and acceptable adverse events (Table 1).
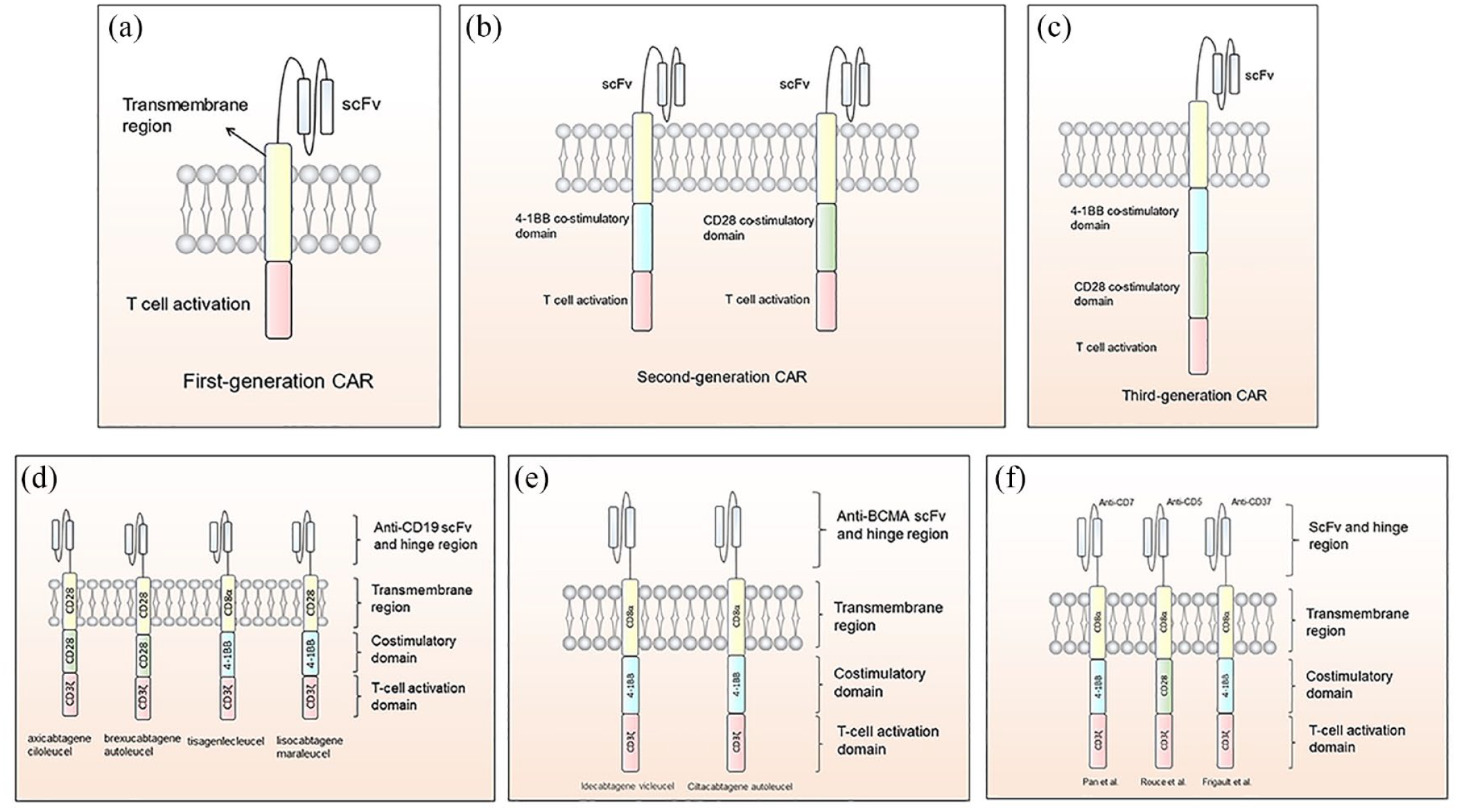
Structure of CARs: (a) The intracellular part of the first-generation CAR comprises a T-cell activation domain and, in most cases, is CD3ζ. (b) Costimulatory domain 4-1BB or CD28 is introduced in second-generation CAR. (c) The third-generation CAR contains both of the molecules. (d) CARs design of the FDA-approved anti-CD19 CAR-T-cell products. (e) CARs design of the FDA-approved anti-BCMA CAR-T-cell product; (f) CARs design of the CAR-T-cells for T-cell malignancies. CAR-T, chimeric antigen receptor T-cell.
The FDA-approved CAR-T products and the major published trials.

B-ALL, B-cell acute lymphoblastic leukemia; BCMA, B-cell maturation antigen; CAR-T, chimeric antigen receptor T-cell; CRS, cytokine release syndrome; DLBCL, diffuse large B-cell lymphoma; FDA, Food and Drug Administration; FL, follicular lymphoma; NTX, neurotoxicity.
Comparison of CAR-T-cells targeting B-cell and T-cell malignancies
The most significant difference in structure between CAR-T-cells targeting B-cell malignancies and T-cell malignancies is the target antigen. CD19 and BCMA molecules are not expressed in T-cells so the CAR-T-cells and normal T-cells would not be recognized. However, the CAR-T-cells targeting T-cell malignancies may also recognize and kill the normal T-cells and CAR-T-cells due to their mutual T-cell antigen. The transmembrane domain usually contains CD28 or CD8α molecules. In most clinical trials for T-cell malignancies, investigators use CAR-T-cells with 4-1BB costimulatory domain [Figure 1(d)–(f); Table 2]. Although there was preclinical study found that 4-1BB-costimulated CAR-T-cells have greater persistence while CD28-costimulated CAR-T-cells have a higher level of cytokines release, 39 more studies are still needed to verify it.
Results of CAR-T therapy clinical trials in T-cell malignancies.

CAR-T, chimeric antigen receptor T-cell; CR, complete remission; CRi, CR with incomplete count recovery; CRS, cytokine release syndrome; GVHD, graft-versus-host disease; HGBCL, high-grade B-cell lymphoma; ICANS, immune effector cell-associated neurotoxicity syndrome; MPAL, mixed phenotype acute leukemia; NR, not reported; ORR, overall response rate; T-ALL, T-cell acute lymphoblastic leukemia; T-LBL, T-cell lymphoblastic lymphoma.
Challenges for CAR-T therapy in T-cell malignancies
These successes inspired investigators to apply this therapy to T-cell malignancies. However, the administration of CAR-T therapy for T-cell malignancies is confronted with several obstacles and challenges 40 (Figure 2).

The challenges of CAR-T therapy in T-cell malignancies: (a) Normal mature T-cells that share mutual antigen with malignant T-cells are lysed after CAR-T infusion, leading to T-cell aplasia. (b) CAR-T-cells that share mutual antigen with malignant T-cells are lysed by themselves and give rise to dysfunction. (c) The contamination of circular malignant cells will eventually cause CAR-T product contamination and impair therapeutic efficacy.
T-cell aplasia and fratricide
Given that potential target antigens should be widely expressed on the T-cell surface, they are also likely expressed on normal T-cells and CAR-T-cells. 40 Thus, after infusion, the CAR-T-cells are directed to not only the tumor cells but also normal T-cells and CAR-T-cells that have the same target antigens on their surface. These cells will be destroyed once they are recognized by the infused cells. The rupture of normal T-cells would give rise to serious T-cell aplasia and immunodeficiency and further cause severe opportunistic infections that could be fatal. 41
The rupture of CAR-T-cells, which is known as fratricide, can seriously hinder the persistence, proliferation, and antitumor efficacy in vivo, leading to unsatisfactory therapeutic efficacy.40,42
Tumor contamination
Moreover, circulating tumor cells exist in peripheral blood; thus, the collection of T-cells could be contaminated with these malignant cells. Circulating malignant cells have a construct and function similar to those of normal T-cells that can also be transduced by exogenous genes. As a consequence, the ultimate CAR-T products would be contaminated with ‘CAR-tumor cells’. Given attempts to inhibit the expression of the target antigen on the CAR-T-cell surface (discussed below) to ameliorate the fratricide, the mixed malignant cells would be modified similarly and subsequently result in antigen escape. This event has been reported in a patient with B-cell leukemia who relapsed by expressing anti-CD19 CAR-tumor cells. The CD19 CAR was transduced in malignant cells in error and led to the combination of CAR and CD19 antigens. This combination prevents CAR-T-cells from recognizing malignant cells and induces antigen escape. 43
Promising target antigens and solutions to the challenges
Choosing an appropriate target antigen for developing CAR-T-cells is extremely significant for this treatment. The ideal target antigen should be expressed on most malignant cells but minimally expressed on normal T-cells and other tissues. We expect that the selection of a target antigen could minimize the adverse events caused by fratricide or on-target off-tumor effects. Herein, we summarize promising target antigens and the outcomes of clinical trials (Table 2). The ongoing clinical trials of these targets are listed in Table 3. In addition, we discuss the probable solution to the aforementioned challenges.
Ongoing clinical trials of CAR T-cell therapy with T-cell antigens.

ALCL, anaplastic large cell lymphoma; ALL, acute lymphoblastic leukemia; AML, acute myeloid leukemia; CAR T, chimeric antigen receptor T-cell; DLBCL, diffusal large B-cell lymphoma; HL, Hodgkin’s lymphoma; NHL, non-Hodgkin’s lymphoma; PMBCL, Primary mediastinal Large B-cell Lymphoma; PTCL, peripheral T-cell lymphoma; T-ALL, T-cell acute lymphoblastic leukemia; CLL, chronic lymphocyte leukemia.
Targets in clinical trials
CD7
Among the numerous latent targets, the most widely studied one is CD7. CD7 is a kind of glycoprotein expressed on the surface of most peripheral blood T-cells, T-cell precursors, and natural killer cells (NK cells).44,45 CD7 is also found in over 95% of T-cell leukemia cases or a subset of PTCLs and previously served as an important clinical marker for diagnosing T-ALL.44,46 Several studies have explored immunotherapy targeting CD7, demonstrating the feasibility of developing CD7 as a therapeutic target in treating T-cell malignancies.47–49 Since it was the most widely studied target, most research on the problems mentioned above was based on CD7.
In 2017, Gomes-Silva et al. 46 proposed that using CRISPR/Cas9 to cut off the CD7 gene before CAR transduction could minimize the fratricide in anti-CD7 CAR-T without hindering the antitumor activity. In their study, CAR-T-cells expressing CD7 failed to expand, but the expansion was restored after removing the CD7 gene. They also demonstrated that the CD7KO CD7 CAR-T-cells (CD7 gene knocked-out CAR-T) maintained potent and specific antitumor activity in vitro. Moreover, they found that CD7KO CD7 CAR-T-cells themselves could still respond to the viral stimulation like non-transduced T-cells, which to some extent alleviates immunodeficiency due to T-cell aplasia. 46 In the same year, Png et al. 50 developed a type of protein expression blocker (PEBL). They combined a CD7-targeted scFv with an endoplasmic reticulum/Golgi-retention motif that confines the newly expressed CD7 to ER/Golgi and successfully reduces CD7 expression on the cell surface of PEBL-CAR T-cells. 50 Compared with mock-transduced T-cells, the fratricide was controlled, and no adverse impact on the T-cell expansion, INF-γ was observed. The PEBL-CD7 CAR-T showed robust cytotoxicity in T-ALL cell lines and murine models of T-ALL. In addition, this technology may be applied soon due to its fitness with existing cell manufacturing processes. 50
In a recent clinical trial, 14 patients with R/R T-ALL were enrolled, including one with Ph + T-ALL, three with ETP-ALL, and five with extramedullary infiltration. These patients had a median of 5 prior lines of therapy. Two patients were treated with a low dose of anti-CD7 CAR-T-cells (0.5×105/kg), eleven with a medium dose (1–1.5×106/kg), and one patient received the highest dose of 2×106/kg. Thirteen of the 14 patients achieved CR or CRi in bone marrow, and 4/5 of the patients with extramedullary infiltration achieved extramedullary remission. CRS occurred in 13 patients but was mild in most patients. 51 In addition, in a study comparing anti-CD7 CAR-T-cells with anti-CD19 CAR-T-cells, the investigators found that the cells targeting CD7 proliferated quickly after infusion, and the duration was longer than that of cells targeting CD19. The anti-CD7 CAR-T-cells did not increase the incidence of adverse event compared with anti-CD19 CAR-T-cells. 33
Given that patients with T-cell malignancies, especially patients with relapsed diseases, usually experience many lines of chemotherapy. This will impair the number and function of normal lymphocytes. Moreover, the circulating malignant cells may contaminate the separated T-cells. It is difficult to collect a sufficient number of qualified T-cells for CAR transduction. Thus, using allogeneic CAR-T-cells for transduction is proposed to solve these problems. Cooper et al. reported a CD7-targeted fratricide-resistant allogeneic CAR-T-cell (UCART7) in 2018. The CD7 and TRAC genes were removed to prevent fratricide and xenogeneic graft-versus-host disease (GVHD), respectively. In preclinical testing, the UCART7 exhibited excellent efficacy in vitro and in vivo without mediating GVHD. 52 Pan et al. 36 conducted a phase I trial of allogeneic anti-CD7 CAR-T-cells that involved 20 patients with R/R T-ALL. CAR-T-cells were infused at doses of 5×105 or 1×106. Ninety percent of the patients achieved CR by the 15th day, and 7 of them accepted allo-HSCT in tandem. At the 6th month of follow-up, the 15 patients were still in remission. All of the patients experienced CRS and cytopenia that were reversible. Twelve patients developed GVHD on the 14th day with a median duration of 3 days. All the symptoms related to GVHD have been alleviated after the administration of methylprednisolone plus ruxolitinib or ruxolitinib alone. However, one patient died of fungal pneumonia at the 5.5th month after infusion. 36
CD5
CD5 is expressed on most T-cell malignancies and normal T-cells.53–55 Mamonkin et al. manufactured an anti-CD5 CAR using CD28 costimulatory domain. In their preclinical testing, the anti-CD5 CAR-T-cells succeeded in eliminating the T-ALL or PTCL cell lines and arresting the progression of the diseases in xenogeneic mice. In addition, a reduction in CD5 expression was noted after CAR transduction, which may explain the transient fratricide observed in their study. 56 Similarly, the downregulation of CD5 expression and mild fratricide were detected in another trial. 57 CD52 was induced into the CAR construct as a switch to control toxicity. Once adverse events occur, the CD52-targeted monoclonal antibody alemtuzumab can be administered to lyse CAR-T-cells carrying CD52. 57 Given the short persistence of CD28-based CAR-T-cells, Mamonkin et al. 58 decided to develop a CAR with a 4-1BB costimulatory molecule that can persist longer. However, increased cell apoptosis was noted in 4-1BB-based CAR-T-cells than in CD28-based CAR-T-cells due to 4-1BB-derived TRAF signaling. Thus, they introduced a Tet-OFF expression system to reversibly regulate CAR expression to alleviate fratricide and elucidated that 4-1BB-based CD5 CAR-T-cells with the Tet-OFF system exhibit increased persistence and antitumor activity compared with CD28-based CD5 CAR-T-cells. 58 Recently, a new CAR structure derived from a fully human heavy chain (FHVH) domain instead of traditional scFv was developed and applied in anti-CD5 CAR-T-cells. The researchers found that FHVH-derived CAR-T-cells have higher degranulation levels and antitumor efficacy than CAR-T-cells with scFv. The VH domain has a smaller size, which is advantageous in recognizing smaller antigenic epitopes. 59
Hill et al. 37 reported a clinical study (NCT03081910) on the treatment of R/R T-ALL and lymphomas with CD5-directed CAR-T-cells. They used autologous peripheral blood mononuclear cells for gammaretroviral transduction and then cryopreserved them. They enrolled 4 T-ALL and 5 T-NHL patients. Three of these patients received CAR-T infusion at a dose of 1×107/m², and the other six patients received 5×107/m². Four to eight weeks later, four patients achieved an objective response, and three patients achieved a complete response. However, two of them relapsed at 6 weeks and 7 months postinfusion. Another patient receiving a second CAR-T infusion and undergoing allo-HSCT in tandem was still in CR at + 125 d of HSCT, suggesting that bridging allo-HSCT after CAR-T infusion may represent a strategy to acquire deeper and longer remission. 37 Three patients experienced CRS during the treatment, and only one of them developed grade 2 CRS requiring tocilizumab. The latest data from this trial demonstrated that cytopenia was the most common side effect, but patients could recover within a month. 60 Feng et al. reported a case with refractory T-cell lymphoma and CNS (Central Nervous System) infiltration. The patient received modified anti-CD5 CAR-T-cells that contained an IL-15/IL-15 sushi complex. They showed that CNS infiltration and CSF (cerebro-spinal fluid) abnormalities were mitigated in patients who had transient T-cell aplasia alone. 61 In general, anti-CD5 CAR-T-cells have good safety and efficacy in the treatment of T-cell malignancies.
TRBC1
One of the dilemmas in treating T-cell malignancy is the poor selectivity mentioned above. Given that normal T-cells and malignant cells may share mutual antigens, CAR-T-cells attack both types of cells after infusion, leading to a severe adverse effect. Unlike B-cell aplasia, it is intolerable for the organism to develop T-cell aplasia. Thus, a more specific target is urgently needed. The T-cell receptor is widely expressed in T-cells and comprises six different polypeptides, including αβ subunits. 62 The β-constant region consists of two different genes called TRBC1 and TRBC2. A single T-cell only expresses each of them, as does the malignant cell. 63 These TCR molecules have similar functions but vary slightly in amino acid sequence; thus, different T-cells can be distinguished. In a normal individual, approximately 35% of T-cells are TRBC1 positive and 65% of T-cells are TRBC2 positive. Targeting either group of cells would not deplete the function of the other group. This feature serves as the cornerstone of anti-TRBC CAR-T therapy 64 (Figure 3). Ideally, the anti-TRBC1 or anti-TRBC2 CAR-T-cells could specifically target malignant cells as well as T-cells that express the corresponding TRBC molecule. Therefore, a large portion of T-cells expressing another TRBC molecule would be spared, and patients could still derive a great response from this approach by avoiding severe fratricide and nonspecific killing of T-cells. Maciocia et al. developed CAR-T-cells targeting TRBC1 and demonstrated that those cells specifically eradicate TRBC1+ cells instead of TRBC2+ cells in vitro and in vivo. Reduced tumor burden and prolonged survival periods were observed in mouse models. 64

The mechanism for anti-TRBC1 CAR-T therapy. Each T-cell expresses a certain TRBC molecule. The CAR-T-cells targeting TRBC1 can specifically distinguish TRBC1+ cells, including tumor cells and normal T-cells. TRBC2+ T-cells are preserved to maintain cellular immunity. CAR-T, chimeric antigen receptor T-cell.
Based on their study, two clinical trials on anti-TRBC1 CAR-T-cells are ongoing. In a trial conducted in Spain and the United Kingdom, a total of 55 patients with T-NHL, such as angioimmunoblastic T-cell lymphoma (AITL), anaplastic large cell lymphoma (ALCL), and peripheral T-cell lymphoma, not otherwise specified (PTCL, NOS) were enrolled and received TRBC1-directed CAR-T-cells infusions at an escalating dose of 25 to 225×106 cells. The trial aims to identify the safety and efficacy of this treatment (NCT03590574). Another trial of anti-TRBC1 CAR-T-cells in patients with R/R TRBC1+ T-cell malignancies, including T-cell leukemia, is currently recruiting (NCT04828174).
CD4
CD4 is expressed in most T lymphocytes, including malignant and normal cells. 65 Due to its rare expression beyond the hemopoietic system, CD4 exhibits the feasibility of serving as a therapeutic target. Pinz et al. conducted preclinical testing using a self-designed anti-CD4 CAR-T-cell (CD4CAR). The KARPAS 299 cells (a cell lymphoma cell line) were lysed after 24 h of coculture with the CD4 CAR, whereas the cells in the control group were still alive. Next, they tested the in vivo activity by injecting CD4CAR into xenogeneic mice and showed that malignancy expansion was arrested. 66 Another recent trial also showed that anti-CD4 CAR-T-cells successfully prevent the proliferation of malignant cells and prolong the survival time of mice with a dose-response relationship. 67 Currently, serval clinical trials are ongoing (Table 3).
The first case reported to undergo treatment with CD4-directed CAR-T-cells was a 54-year-old patient with Sezary syndrome. Before the CAR-T infusion, he had been resistant to prior lines of chemotherapy, and tumor infiltration was detected in his skin. The patient received CAR-T infusion at a dose of 3×106/kg. On Day + 13 after infusion, the patient achieved CR. On Day + 28, the biopsy demonstrated that the skin was free from tumor infiltration. Moreover, no grade II CRS or other adverse events were observed during his treatment. 68 Targeting CD4 is effective but needs to be more cautious. Given on-target effects, patients may still develop serious CD4+ cell aplasia that resembles HIV. The solution to this issue includes shortening the lifetime of CAR-T-cells or using NK-cells for CAR transduction as their lifespan is shorter. 69
CD30
CD30 is a transmembrane receptor of the TNFR superfamily and is mainly expressed on Hodgkin’s lymphoma (HL), anaplastic large cell lymphoma (ALCL), and approximately 1/3 of T-ALL.70–72 ALCL is characterized by uniformly expressed CD30. Moreover, it was also reported that CD30 expression was increased during high-dose chemotherapy in T-ALL. 72 Thus, CD30 was deemed a latent target for immune therapy of T-cell malignancies. Brentuximab vedotin (BV) is a flourishing antibody-drug conjugate. An anti-CD30 antibody binds to a cytotoxic agent monomethyl auristantin E (MMAE) that can disrupt microtubules to inhibit tumor cells. 73 Several published clinical trials of BV in patients with R/R ALCL showed that the outcomes were ameliorated using this approach. In a phase 2 study, the OS was as high as 79% with a median response duration not reached.74,75 These results suggested that CD30 may also represent an excellent CAR-T target, and its feasibility has been demonstrated in a previous preclinical testing. 76
In 2017, Ramos et al. 77 described a clinical trial that involved two ALCL and seven R/R HL patients. The two ALCL patients were diagnosed with cutaneous ALK− ALCL and systemic ALK+ ALCL, separately. The patient with ALK+ ALCL received anti-CD30 CAR-T-cells (CD30. CAR-Ts) at a dose of 2×108/m² and presented PR by PET/CT in the 6th week postinfusion. Furthermore, this patient received three additional infusions later and achieved CR that lasted for 9 months. However, no response was observed in the other patient with ALK- ALCL. 77 Regarding safety, the nine patients enrolled in this trial did not demonstrate any adverse reactions related to CRS or CD30 CAR-Ts. 77 In another open-label phase I trial conducted by Wang et al., the only patient diagnosed with cutaneous ALCL received CAR-T-cells at a dose of 2×107/kg. The patient underwent several lines of treatment before the infusion, including chemotherapy, BV, radiation therapy, and surgery. He achieved PR after the first infusion, and the mass on the skin disappeared after receiving the second infusion 4 weeks later. Adverse events mainly presented as increased levels of serum liver enzyme and γ-GGT. 78 In 2018, Wang et al. demonstrated that in a trial involving four patients with HL and two patients with ALCL, five of them achieved CR after anti-CD30 CAR-T infusion (including the two ALCL patients). Five patients experienced grade 0–1 CRS, but one patient with HL died of grade 4 CRS. 79 In addition, Voorhees et al. reported a case with refractory/relapsed CD30 + enteropathy-associated T-cell lymphoma (EATL). The patient received standard lymphodepletion chemotherapy before receiving anti-CD30 CAR-T dose of 2×108/m². He experienced grade 1 CRS on the 12th day after infusion and recovered on the 15th day. In the sixth week, the patient achieved PR, and the previously affected lymph nodes all disappeared as assessed by PET/CT scan. At the 24-month follow-up, the patient maintained continued remission. 80 These findings indicated that anti-CD30 CAR-T-cells have good safety and efficacy. It has great potential for the treatment of ALCL. However, there were still cases who died of severe CRS. It suggests that we need to better select the timing and dose of treatment. As a consequence, more data from diverse clinical trials are necessary to further evaluate this novel treatment.
CD37
CD37 is a transmembrane protein of tetraspanin superfamily. It mainly distributes on B-cells and partial T-cell lymphomas. 81 For CD37 + B-lymphomas and CLL, a variety of monoclonal antibody-based drugs targeting CD37 have been assessed in clinical trials and have preliminarily revealed excellent safety and efficacy.82–84 These findings suggest that CD37 could act as a therapeutic target. Scarfò et al. 85 developed an anti-CD37 CAR-T-cell (CAR-37) with 4-1BB costimulatory domain. They verified that CD37 was expressed in PTCL cell lines and cultivated the PTCL cells with CAR-37 together in vitro. After coculture, the cell lines Hut78 and Fepd were eliminated, and no obvious fratricide was observed, indicating that CAR-37 has antitumor activity against PTCL cells. 85 Given that not all T-cell lymphomas express CD37, testing CD37 expression before therapy is necessary to select suitable patients.
The only ongoing trial of CD37-directed CAR-T-cells (NCT04136275) planned to enroll patients with R/R hematologic malignancy, including leukemia, B-lymphoma, and T-lymphoma, to evaluate the safety at a starting dose of 100×106 CAR-T-cells. Four patients received anti-CD37 CAR-T infusion, including two with high-grade B-lymphoma, one with HL, and one with cutaneous T-cell lymphoma (CTCL) in a phase I trial. The patient with CTCL received 19×106 CAR-T-cells due to poor ex vivo proliferation, and he achieved CR on the 28th day postinfusion. All four patients had detectable in vivo CAR-T-cells proliferation and experienced CRS and immune effector cell-associated neurotoxicity syndrome (ICANS). Of note, two patients receiving a CAR-T dose of 100×106 CAR-T-cells developed prolonged pancytopenia with marrow aplasia that required allo-HSCT. 38
Other targets
CD1a
CD1a is a molecule expressing exclusively in cortical thymocytes or Langerhans cells, making it a good target for treating cortical T-ALL and Langerhans cell histiocytosis. 86 CD1a is limitedly expressed on developing cortical thymocytes but is not expressed in other types of T-cells; thus, it may represent an effective method to prevent fratricide and immune suppression in CAR-T therapy. Sánchez-Martínez et al. 87 showed that the CAR-T-cells with anti-CD1a scFv could expand without T-cell fratricide and was cytotoxic against CD1a cell lines. CD1a-redirected T-cells proliferated up to 200 times on the 12th day in vitro, which was similar to that noted for MOCK T-cells. In the xenogeneic cortical T-ALL model, the infused CD1a-directed T-cells decreased the tumor burden. However, Leong et al. 88 found that CD1a is rarely expressed in R/R T-ALL. In most cases, CD1a+ is a marker associated with a good prognosis, and patients usually do not need to receive CAR-T infusion. Thus, the suitable patients for this treatment are sparse. In addition, it is only suitable for patients with cortical T-ALL, which further limits the application of anti-CD1a CAR-T products. There is currently no ongoing clinical trial for CD1a CAR-T therapy.
CD21
CD21 is regarded as a pan-B antigen expressed on normal B-cells, playing an important role in innate and specific immune responses. 89 CD21 binds to CD19/CD81 on the surface of B-cells to form a B-cell coreceptor, by which B-cells enhance their sensitivity and activation. 90 A previous study demonstrated that CD21 was expressed on T-ALL cells and was related to NOTCH signaling in T-ALL,89,91 bringing it to the attention of investigators. Maciocia et al. 92 found that CD21 was extensively expressed on T-ALL cells and normal B-cells, whereas it was minimally expressed on mature normal T-cells. Hence, developing CD21 as a target is a promising method to overcome fratricide and on-target off-tumor effects. Recently, in murine models of T-ALL, anti-CD21 CAR-T-cells demonstrated superior performance in reducing tumor burden and exhibited increased OS compared with anti-CD19 CAR-T-cells. 92 Given the restricted expression of CD21 on B-cells and malignant T-cells, lethal complications caused by severe immunodeficiency are less likely to develop. The downside of CD21 is that only a portion of malignant cells express CD21.89,92 Thus, antigen escape occurs at some point, and the patient ultimately relapses.
CCR4
Chemokine receptor 4 (CCR4) is mainly expressed by Th2 cells, Treg cells, and skin lymphocyte antigen-positive homing T-cells. 93 CCR4, with its ligands CCL17 and CCL22, encourages the accumulation of CCR4-expressing T-cells in the skin and the migration of Treg cells to the tumor microenvironment. This mechanism is important in the pathogenesis of CTCL. 94 CCR4 expression is upregulated in CTCL, MF, SS, and ATLL.93,95,96 Currently, a humanized monoclonal antibody against CCR4 mogamulizumab has been approved for the treatment of R/R CTCL, MF, SS, and ATLL.97,98 This finding indicates that it is a potential target for CAR-T therapy.
Perera et al. designed an anti-CCR4 CAR construct with 4-1BB costimulatory molecule. They demonstrated the cytotoxicity of the CCR4-directed cells toward CTCL cell lines and noted that CCR4-targeted CAR-T-cells will not cause severe disorder in the T-cell subset. 99 However, several adverse events resembling those of mogamulizumab, including thrombocytopenia and toxic epidermal necrolysis, were noted. 99 Thus, more research on its safety and efficacy is still required before its application in patients.
CCR9
CCR9 expression is limited to immature T lymphocytes and intestinal cells. CCR9 is expressed in greater than 70% of T-ALL cases and less than 5% of normal cells.100,101 CCR9 promotes the migration, infiltration, and expansion of leukemia cells when it binds to its ligand CCL25.101,102 Somovilla-Crespo et al. 103 generated a type of anti-CCR9 antibody and demonstrated that it inhibits the growth of CCR9+ leukemia cells in a mouse model. Recently, Maciocia et al. designed a CAR-T-cell with anti-CCR9 scFv, and the anti-CCR9 CAR-T-cells showed excellent antitumor efficacy in their study. Moreover, it effectively alleviated fratricide and T-cell aplasia for its limited expression in normal tissue. 100
Solutions to overcome the current challenges and enhance efficacy
Solutions to T-cell aplasia
Patients with T-cell aplasia are predisposed to opportunistic infections that are sometimes fatal. Unlike the injection of immunoglobulin for B-cell aplasia caused by anti-CD19 CAR-Ts, an similar effective approach to T-cell aplasia has not been developed. Thus, taking precautions against T-cell aplasia is important. (1) Choosing a highly specific target to manufacture CAR-T products is the basic measure to avert T-cell aplasia. The ideal target should be expressed exclusively on malignant cells and rarely expressed on normal T-cells such that CAR-T-cells can identify the tumor precisely. One of the examples involves targeting the TRBC previously mentioned. The CAR-T-cells are transduced with scFv targeting TRBC1 or TRBC2, and these CAR-T-cells would only kill either cell population (TRBC1+ cells or TRBC2+ cells; Figure 3). 64 (2) Introducing the suicide gene as a ‘switch’ to regulate the CAR-T-cells is also an effective method. Researchers have incorporated the inducible caspase-9-based suicide gene as suicide gene in CAR.104,105 By activating the suicide gene, CAR-T-cells could be eliminated quickly when unwanted adverse events occur. (3) Using logic gates (AND gate) technology may be useful to reduce adverse effects of CAR-T-cells (Figure 4). 106 Roybal et al. introduced the synNotch receptor in CAR-T-cells. When binding an antigen, the synNotch receptor releases a transcription factor to activate the expression of another CAR that targets a different antigen. The CAR-T-cells only function when both of the antigens are recognized. This feature enhances the specificity of CAR-T-cells, but sufficient target antigens must be identified. 107 However, enhanced safety is typically accompanied by impaired efficacy. It is important to strike a balance between safety and efficacy. Moreover, bridging allo-HSCT after CAR-T infusion is necessary if serious T-cell aplasia has occurred. 108

Solutions to overcome the current challenges: (a) T-cell aplasia. (b) Fratricide. (C) Tumor contamination.
Solutions to fratricide
Fratricide causes CAR-T-cells to recognize and lyse each other, which exerts a negative effect on the activity, proliferation, and survival of CAR-T-cells. (1) Finding a target antigen expressing on malignant cells while not on therapeutic CAR-T-cells. For instance, CD1a is a target antigen expressed mainly on developing cortical thymocytes but rarely expressed in other stages of T-cells. 87 The expression of CD5 on CAR-T-cell is downregulated after CD5 CAR-T-cell infusion. 56 (2) Inhibition of the target antigens expression on CAR-T-cells will help reduce fratricide. Knocking out TCR or the gene of target antigen by genome editing may greatly ameliorate fratricide. 109 A study showed that the CRISPR/Cas9-mediated TCR knockout eliminated the alloreactivity of allogenic CD19 CAR-T-cells. 110 It is possible that this type of technology would be effective in fratricide. For CD7 CAR-T-cells, the CD7 gene was knocked out in CAR-T-cells or PEBL technology/anti-CD7 antibody were employed to prevent the mutual recognition of CD7.46,50,111 The inhibition of CD7 expression successfully eliminates fratricide. (3) Using the cells without mutual antigen expressed on its surface, such as NK-cells. 112 It has been demonstrated that NK-cells have similar efficacy to T-cells in killing malignant cells. 113 To date, the NK-92 cell line has shown antitumor activity and has been used to develop CAR products with CD4 and CD5 as targets.69,114,115 However, NK-cells do not exhibit long-term viability when the cytokine is absent. 116 (4) Novel approaches to fratricide, such as using nanobody-derived CAR-T-cells or naturally selected fratricide-resistant CAR-T-cells, have demonstrated superiority in the treatment of CD7+ malignancy (Figure 4).34,117 They provided a new way to overcome fratricide excluding gene editing, which may help maintain the natural characteristics of cells and enhance the safety as well as efficacy. Liu et al. cultivated anti-CD7 CAR-T-cells in vitro and selected cells that survived after fratricidal natural selection. In their study, this type of CAR-T products presented better cytotoxicity and resistance to fratricide than CAR-T-cells edited by CRISPR/Cas9. The naturally selected CAR-T-cells (NS7CAR) decreased the tumor burden in mouse models in the first 2 weeks after infusion. 118 In their later phase I clinical trial, NS7CAR was applied to 20 patients (14 with T-ALL and 6 with T-LBL). By Day 28, 94.12% (16/17) of patients with bone marrow infiltration achieved MRD negative CR, and 55.6% (5/9) of patients with extramedullary infiltration achieved extramedullary CR with a median time of 29 days. Most patients did not experience severe CRS (grade ⩽ 2) except 1 patient who developed grade 3 CRS. 34 In another study, Chen et al. used an anti-CD7 nanobody fragment connected to an ER/Golgi retention domain to manufacture CAR-T-cells (CD7ΔCD7-CAR-T-cells). These researchers demonstrated that nanobody-derived CAR-T-cells can avoid fratricide and have a great antitumor effect. 119 Zhang et al. 117 reported a clinical trial with this kind of anti-CD7 CAR-T-cells. Of the eight patients enrolled in the trial, the CR rate at 3 months was 87.5% (7/8), and no life-threatening CRS or ICANS was observed.
Solutions to tumor contamination
Given that T malignant cells are derived from normal T-cells or their precursors, they are similar to each other. Thus, it is often prone to incorporate the malignant cells into collected T-cells. One way to avoid contamination involves the use of NK-cells that are easier to distinguish. The markers of NK-cells are different from T-cells. 120 Therefore, NK-cells are less likely contaminated by T malignant cells. Another solution to this problem involves the use of T-cells from allogeneic donors (Figure 4). However, the allogeneic CAR-T-cells may give rise to intolerable GVHD or be eliminated by immune system, which impairs the efficacy of this therapy. 121 Cooper et al. reported the feasibility of an allogeneic anti-CD7 CAR-T product UCART7. It showed satisfactory antitumor efficacy in vitro and in vivo without mediating GVHD. 52 Pan et al. initially administered the allogeneic anti-CD7 CAR-T-cells to patients with R/R T-ALL. The result of their clinical trial demonstrated that the infused cells effectively proliferated and the CR rate of enrolled patients was up to 90%. Although some patients developed GVHD, they were mild and reversible. 36 Moreover, given that the transduction efficiency and vector copy number of CAR-T-cells can be detected and evaluated by flow cytometry and qPCR, improvements in these technologies may be helpful to this problem. 122
Other solutions to enhance CAR-T efficacy
For the CAR-T therapy for B-cell malignancies, investigators have explored a variety of methods to enhance the efficacy, such as combination strategies and dual-target CAR-T-cell therapy.123,124
A preclinical study showed that the expression of PD-1 and PD-L1 was upregulated after CAR-T infusion, which will inhibit the effector function of CAR-T-cells. However, the injection of anti-PD-1 antibody enhanced the accumulation of CAR-T-cells and the cytokine secretion. 125 And the enhanced efficacy was observed in a clinical trial that combined anti-CD19 CAR-T-cells and Nivolumab. 126 This suggests that combination therapy may similarly improve the efficacy of CAR-T-cells in the treatment of T-cell malignancies. However, there is no such study in the treatment of T-cell malignancies and more studies are required to confirm it.
Recently, Dai et al. developed a kind of CD5/CD7 bispecific CAR-T-cell to treat R/R T-cell malignancies. Although the dual-target CAR-T-cells can mitigate tumor antigen escape, targeting multiple antigens will give rise to more severe T-cell aplasia and fratricide. Thus, they knocked out the CD5 gene and CD7 gene before CAR transduction to prevent fratricide. In their study, the CD5/CD7 bispecific CAR-T therapy showed satisfactory antitumor efficacy in mouse model. 127 This may be an effective way of improving CAR-T therapeutic efficacy but it needs clinical trials to verify its safety in humans.
Conclusion and future direction
The development of CAR-based immunotherapy is a landmark in the treatment of hematological malignancies. Compared with B-cell malignancies, T-cell malignancies are associated with greater heterogeneity and worse prognoses. Distinctive challenges of CAR-based treatment in T-cell malignancies have been noted. Severe T-cell aplasia caused by the on-target off-tumor effect, CAR-T-cells fratricide, and contamination of malignant cells represent urgent problems that need to be solved. Selecting target antigens with increased specificity, introducing the suicide gene as a ‘switch’, removing the target genes by gene editing, and using NK-cells or donor-derived T-cells for CAR transduction are current solutions to these problems. To date, many targets under development have demonstrated good efficacy in preclinical testing. However, given the safety and applicability, only a few of these targets entered the clinical trial stage. CD7 is the most widely studied target at present. It exhibited impressive efficacy and safety of both allogeneic and autologous CAR-T products in phase I studies, and a phase II trial of allogeneic anti-CD7 CAR-T-cells is ongoing. 36 The TRBC1 or TRBC2 gene is only expressed on half of the T-cells, so these genes might be a good target to overcome T-cell aplasia and fratricide. 64 CD21 and CCR9 are newly proposed target antigen that are restricted to malignant T-cell compartments, which may help to avoid the T-cell aplasia and fratricide. Some patients underwent the cytokine release syndrome in diverse trials. However, the toxicity was reversible and transient in most cases. The main work in the future is still to find solutions to overcome the obstacles mentioned above. The combination therapy and dual-target CAR-T are potential way to enhance the efficacy. The development of this treatment is still at an early stage. Thus, more research are still needed for the approval of this treatment.