
Abstract
Chemotherapy resistance and relapse remain significant sources of mortality for children and adults with acute myeloid leukemia (AML). Further intensification of conventional cytotoxic chemotherapy is likely not feasible due to the severity of acute and long-term side effects upon normal tissues commonly induced by these drugs. Successful development and implementation of new precision medicine treatment approaches for patients with AML, which may improve leukemia remission and diminish toxicity, is thus a major priority. Tumor antigen-redirected chimeric antigen receptor (CAR) T-cell immunotherapies have induced remarkable responses in patients with relapsed or chemorefractory B-lymphoblastic leukemia, and similar strategies are now under early clinical study in adults with relapsed/refractory AML. However, potential on target/off tumor toxicity of AML CAR T-cell immunotherapies, notably aplasia of normal myeloid cells, may limit broader implementation of such approaches. Careful selection of optimal target antigens, consideration of toxicity mitigation strategies, and development of methodologies to circumvent potential CAR T-cell resistance are essential for successful implementation of cellular immunotherapies for patients with high-risk AML.
Keywords
Introduction
Acute myeloid leukemia (AML) is the most common leukemia occurring in adults and the second most common leukemia of childhood. Intensive multiagent chemotherapy regimens for patients with AML have changed little during the past four decades and are associated with considerable acute and chronic toxicities. Despite consolidation with hematopoietic stem cell transplantation (HSCT) for those with high-risk AML, relapse-free and overall survival for many patients remains poor.1–5 New therapeutic approaches are compulsory to improve outcomes.
Targeted antibody-based or cellular immunotherapies are particularly attractive for patients with demonstrated chemotherapy-resistant disease, who often have few remaining curative treatment options. Studies have demonstrated improved outcomes for subsets of patients with high-risk AML treated with chemotherapy and the CD33-targeted antibody–drug conjugate (ADC) gemtuzumab ozogamicin,6–8 which led to approval of gemtuzumab by the United States Food and Drug Administration (FDA) and the European Medicines Agency in adults and children with de novo or relapsed AML. Early phase clinical trials of other antibody-based therapeutics, including new CD33 and CD123 [interleukin (IL)-3Rα] targeting ADCs and bispecific T-cell engager (BiTE)/dual affinity retargeting (DART) antibodies are currently underway in adults with relapsed/refractory AML.9–14
Particular progress has recently been made with adoptive cellular therapies using autologous or allogeneic T cells engineered with synthetic chimeric antigen receptors (CARs) redirected against tumor antigens with remarkable early-phase clinical trial results in patients with B-lymphoblastic leukemia (B-ALL) treated with CD19 or CD22 CAR T cells.15–22 The logistics and mechanics of CAR T-cell engineering for patients with acute leukemias and potential safety modifications have been delineated in numerous recent reviews.23–28 In contrast to earlier T-cell receptor (TCR)-directed T cells, genetically engineered CAR T cells (usually autologous and permanently modified via retroviral or lentiviral transduction) bind to cell surface antigens without the need for traditional matching of major histocompatibility complex (MHC) antigens to prevent alloimmunization. Upon binding of the synthetic CAR to its target antigen, intracellular signaling via costimulatory domains induces T-cell activation and marked expansion, often resulting in rapid and complete cancer cell cytotoxicity. However, ‘on target/on tumor’ sequelae of CAR T-cell activation and proliferation can result in life-threatening toxicities, including neurologic dysfunction, cytokine release syndrome (CRS), and macrophage activation syndrome.29–33 Concomitant ‘on target/off tumor’ effects of CAR T cells caused by indiscriminate cellular binding to the same antigens on nonmalignant normal cells can also be quite detrimental to the host and have been described in detail elsewhere.24,34–36
In patients with AML, hematologic toxicity with potential CAR T-cell-induced myeloablation is a legitimate particular concern given the lack of currently known AML-only surface proteins and expression of targeted candidate antigens on normal myeloid precursor cells (Figure 1). As such, rescue of CAR T-cell-treated patients with HSCT to restore normal myelopoiesis may be required. High potency CAR T-cell exhaustion 37 and immune escape with target antigen loss or immunophenotype switching38–40 are also emerging as major mechanisms of resistance to CAR T-cell and antibody-based immunotherapies, a reproducible lesson now well learned from treated patients.19,41 This review discusses the current bench-to-bedside landscape of antigen-redirected CAR T-cell immunotherapies for patients with AML, continued challenges in the field, and emerging strategies that may optimize therapeutic efficacy while reducing potential toxicity.

Balancing efficacy and toxicity of chimeric antigen receptor (CAR) T-cell immunotherapy for acute myeloid leukemia (AML). CNS, central nervous system.
Particulars of AML CAR T-cell development: picking target antigens, potency, persistence, and potential problems
Prolonged B-cell aplasia is an expected (and perhaps desired) bystander toxicity in patients treated with CD19 CAR T cells given the concomitant presence of CD19 on normal B cells.16,17,42 Patients with B-ALL treated with these targeted immunotherapies are now monitored closely for continued B-cell loss as a biomarker of CD19 CAR T-cell persistence. Surprisingly, few untoward effects of this toxicity have been observed to date, as CD19 expression is restricted to B cells, and patients with continued B-cell depletion and resultant hypogammaglobulinemia can be safely supported with monthly intravenous immunoglobulin infusions to minimize infectious complications. 43 Similar B-cell aplasia has now been observed in patients with relapsed/refractory B-ALL treated with the CD22 ADC inotuzumab ozogamicin or CD22 CAR T cells given similar levels of CD22 expression on normal B lymphocytes.19,44,45
Ideal AML surface antigen characteristics for successful immunotherapeutic targeting include restriction to malignant myeloblasts without expression on normal hematopoietic stem cells or on vital normal nonhematopoietic tissues. Preferably, the antigens should be critical to leukemogenesis initiation or maintenance or expressed on leukemia-initiating cells to maximize AML eradication versus expressed only on more mature, often clonally heterogeneous bulk leukemia cell populations. Given the lack of AML cell-only antigens identified to date, most preclinical and clinical immunotherapy efforts have instead attempted to identify a therapeutic window of targeting myeloid antigens overexpressed on AML blasts that are present at lower or minimal levels on normal tissues. It is thus possible, even probable, that permanently modified AML CAR T cells under current study will eradicate normal antigen-expressing myeloid progenitor cells or induce significant myeloablation. As many AML-associated surface proteins of interest for CAR T-cell targeting are not restricted to the hematopoietic compartment (e.g. CD33 on hepatic Kupffer cells, CD44 on keratinocytes, CD123 on endothelial cells, CD135 on neural and testis tissues), risk of appreciable nonhematologic side effects must also be carefully considered. Optimal AML immunotherapy approaches should thus aim when possible to target surface antigens occurring only in the hematopoietic compartment or those where ‘bystander’ toxicity to antigen-expressing normal tissues will be minimal or clinically tolerable.
To address the issue of leukemia cell antigen specificity, a recent transcriptomic and surfaceome proteomic analysis profiled human AML cell lines and primary patient samples and reported unique pairings of antigens detected on AML blasts, but not co-occurring on normal hematopoietic cells. The investigators suitably concluded that dual antigen-cotargeted CAR T cells will likely induce less toxicity upon normal hematopoietic and non-hematopoietic tissues in which only single antigen expression occurs. 46 Another recent preclinical study demonstrated greater in vivo anti-leukemia activity and prolonged animal survival in AML cell line xenograft models treated with compound CD33xCD123-targeting CAR T cells versus single antigen-redirected T cells. 47 These combinatorial therapeutic strategies to increase efficacy and potentially reduce toxicity are analogous to that of bispecific CD19xCD22 CAR T cells 48 under current clinical evaluation in adults and children with relapsed B-cell ALL [ClinicalTrials.gov identifier: NCT03241940].
While antecedent experience with and demonstrated safety of monoclonal antibody-based immunotherapeutics provides some reassurance that targeting of the same antigens with CAR T cells will be similarly well tolerated, it is imperative to remember that these ‘living drugs’ are likely far more potent and longer lasting than their antibody counterparts, thereby increasing potential for both increased anti-AML efficacy and prolonged toxicity. This paradox must be carefully balanced: CAR T-cell persistence for some duration is unquestionably necessary to eradicate leukemia, yet potential ensuing myeloablation caused by such therapies poses great hematologic and infectious risk to the host without additional intervention. As such, investigators focused on AML CAR T-cell development have appropriately attempted to maximize efficacy and minimize toxicity via subsequent T-cell termination, or rescue with allogeneic HSCT to restore hematopoiesis.36,49–51
Preclinical studies of AML CAR T-cell immunotherapy
In the past decade, numerous preclinical studies have reported promising in vitro and in vivo activity of CAR T cells targeting various myeloid antigens in human AML cells and patient-derived xenograft (PDX) models, including Lewis-Y, 52 CD33,53–59 CD123,35,47,50,51,54,60–64 CD44v6, 49 the folate receptor β, 65 CD38,66,67 and the FLT3 receptor (CD135),68–70 and CLL-1 (CLEC12A).71–73 All of these targeted CAR T cells have demonstrated potent in vitro cytotoxicity and cytokine production when incubated with antigen-positive AML cell lines or primary cells (Table 1). In many of these studies, a single dose of permanently modified CAR T cells also completely eradicated in vivo human AML cell proliferation in engrafted immunocompromised mice.
Candidate AML antigens for CAR T-cell immunotherapy and data from preclinical studies.
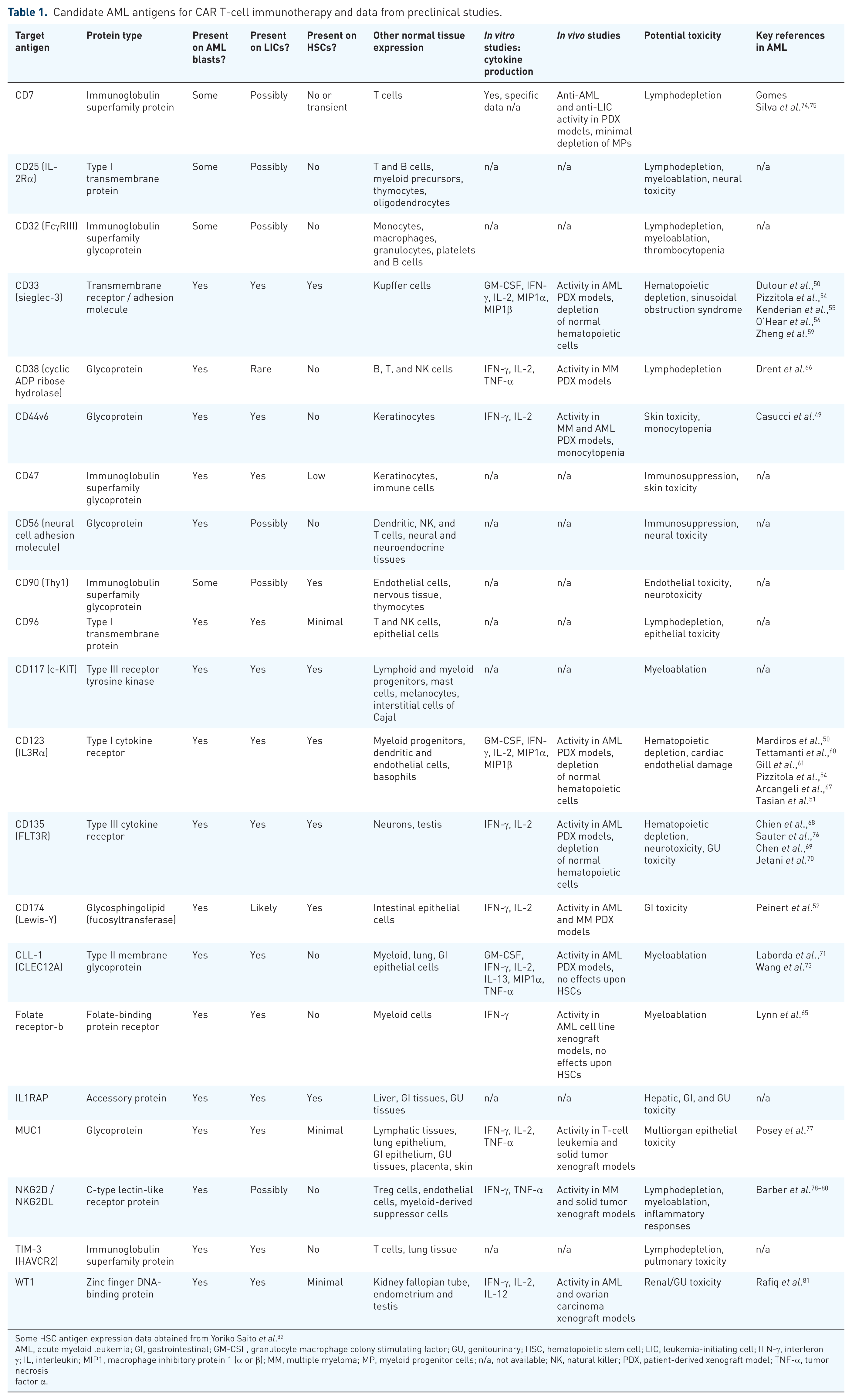
Some HSC antigen expression data obtained from Yoriko Saito et al. 82
AML, acute myeloid leukemia; GI, gastrointestinal; GM-CSF, granulocyte macrophage colony stimulating factor; GU, genitourinary; HSC, hematopoietic stem cell; LIC, leukemia-initiating cell; IFN-γ, interferon γ; IL, interleukin; MIP1, macrophage inhibitory protein 1 (α or β); MM, multiple myeloma; MP, myeloid progenitor cells; n/a, not available; NK, natural killer; PDX, patient-derived xenograft model; TNF-α, tumor necrosis factor α.
However, several groups have also reported potent in vivo hematologic toxicity due to T-cell persistence in AML PDX models,49,50,61,76 highlighting potential necessity of T-cell termination strategies to decrease toxicity to normal tissues. Such approaches include incorporation of suicide genes in the CAR construct for subsequent chemical- or antibody-mediated T-cell termination, CAR T-cell depletion with monoclonal antibodies directed against T-cell surface proteins, and mRNA CAR-electroporated T cells that last for only a few days in vivo. 36 Each of these strategies has particular advantages and disadvantages. The suicide switches herpes simplex virus thymidine kinase (HSV-tk) and inducible caspase 9 (iC9) can induce rapid, but potentially incomplete, CAR T-cell death following administration of the antiviral medication ganciclovir or chemically inert inducer of dimerization (CID) prodrugs, respectively. The HSV-tk strategy was initially developed to control T-cell alloreactivity in graft-versus-host disease following HSCT 83 and has been integrated into some preclinical CAR T-cell studies, 49 but has proven to be fairly immunogenic in patients. Conversely, the iC9 approach with administration of the CID rimiducid (AP1903) is minimally immunogenic and does not depend upon cell division to induce apoptosis, which may be more clinically desirable.66,84,85 Several groups have also incorporated coexpression of synthetic surface proteins [e.g. truncated epidermal growth factor receptor (tEGFR), CD20] in CAR T cells and demonstrated potent T-cell depletion following administration of specific monoclonal antibodies, such as anti-EGFR cetuximab or anti-CD20 rituximab. Administration of the anti-CD52 antibody alemtuzumab has also been used to eliminate CAR T cells via targeting of endogenously occurring cell surface CD52.47,51 Use of ‘biodegradable’ RNA CAR T cells is another attractive approach given their self-limited in vivo lifespan and thus lower risk of inducing sustained off tumor toxicity.51,55,86 However, RNA CAR T cells also have limited expansion potential that is likely required for sustained leukemia cure, and administration of multiple cell infusions is likely necessary to achieve initial remission. Based on promising preclinical data, the tEGFR/cetuximab, CD20/rituximab, alemtuzumab, and iC9/AP1903 approaches have been incorporated into clinical CAR T-cell products under current investigation in adults with relapsed/refractory acute leukemias,18,20,87,88 although actual depletion of CAR T cells in patients with leukemia via these methods has not been reported.
Phase I clinical trial testing of AML CAR T-cell immunotherapy
Despite the recent explosion of preclinical studies demonstrating exciting cytotoxic potential of various AML antigen-targeting CAR T cells, translation to the clinic has been much slower and highlights inherent challenges and risks in developing these treatment strategies for patients with AML. To date, a small number of adults have been treated with AML CAR T-cell immunotherapies via early phase clinical trials (Table 2).
Current phase I clinical trials of CAR T-cell immunotherapy for patients with relapsed/refractory AML.
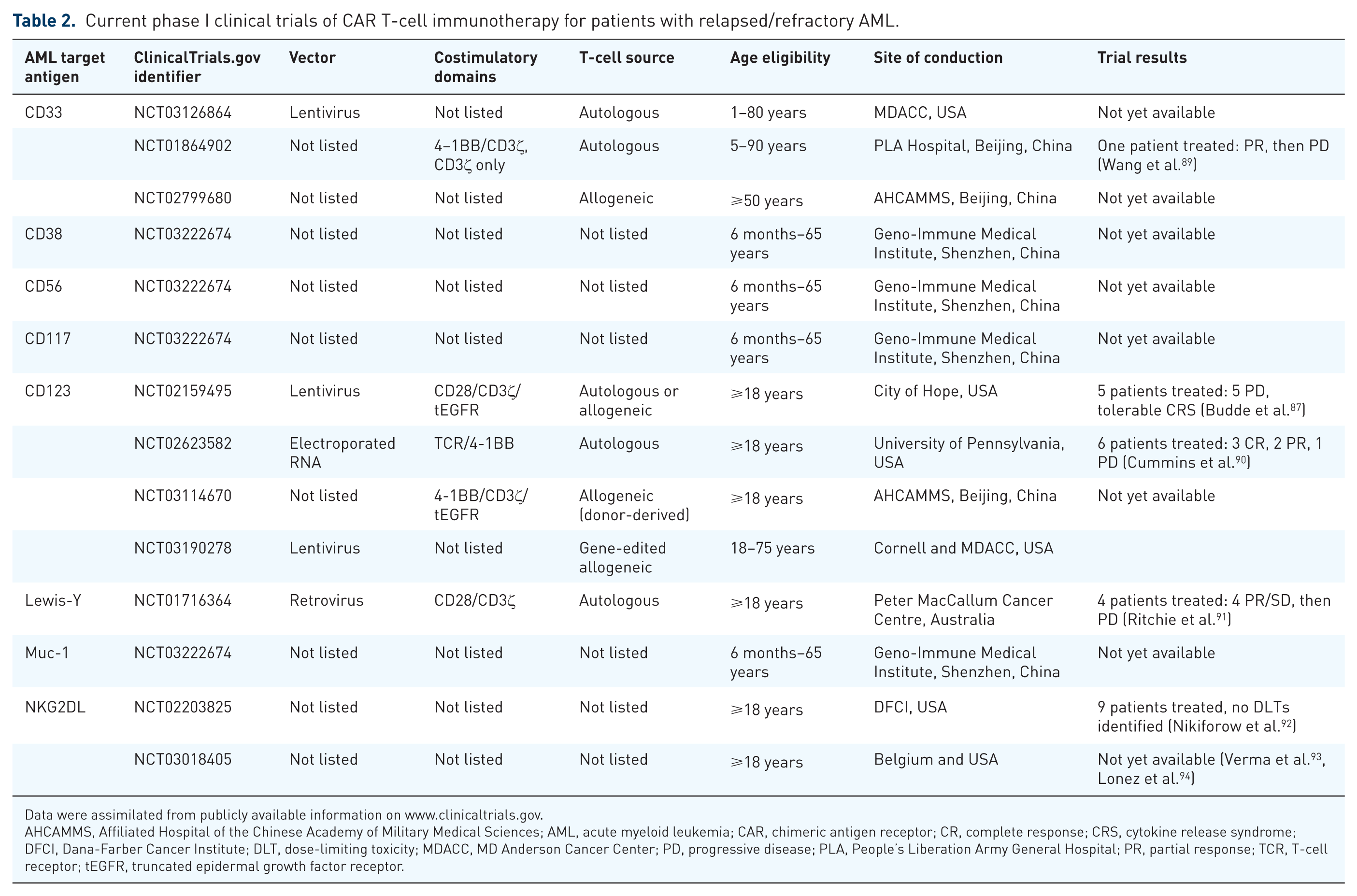
Data were assimilated from publicly available information on www.clinicaltrials.gov.
AHCAMMS, Affiliated Hospital of the Chinese Academy of Military Medical Sciences; AML, acute myeloid leukemia; CAR, chimeric antigen receptor; CR, complete response; CRS, cytokine release syndrome; DFCI, Dana-Farber Cancer Institute; DLT, dose-limiting toxicity; MDACC, MD Anderson Cancer Center; PD, progressive disease; PLA, People’s Liberation Army General Hospital; PR, partial response; TCR, T-cell receptor; tEGFR, truncated epidermal growth factor receptor.
A phase I study conducted at the Peter MacCallum Cancer Centre in Melbourne [ClinicalTrials.gov identifier: NCT01716364] treated four adult patients with AML with a single dose of radioactively labeled second-generation CAR T cells redirected against Lewis-Y (LeY), a carbohydrate antigen overexpressed on many cancer types, including AML. 91 Three patients had minimal residual disease (MRD)-level marrow involvement at study entry, while the fourth patient had active AML with 70% marrow involvement at the time of T-cell infusion with up to 1 × 109 cells at 14–38% transduction efficiency. Single photon emission computed tomography (SPECT) imaging of infused patients and quantitative polymerase chain reaction (PCR) monitoring of peripheral blood transgene levels demonstrated initial pulmonary trafficking of T cells with subsequent localization to spleen and bone marrow. While transient responses (range 1–23 months) with stable disease or leukemia reduction were observed in the four patients, all eventually relapsed. Importantly, this initial trial provided first clues about cytokine release syndrome (CRS) as a potential biomarker of CAR T-cell-induced responses, as the patient with high-level marrow involvement treated with anti-LeY cells experienced pyrexia, rigors, and skin rash exacerbation with biopsy-proven lymphocytic infiltrates at approximately 1 week post T-cell infusion. These studies also demonstrated PCR-detectable persistence of CAR T cells in vivo lasting several months. 91
A current phase I trial in Beijing [ClinicalTrials.gov identifier: NCT01864902] is assessing the safety of second-generation CD33-redirected CAR T cells in patients with relapsed/refractory AML. The investigators reported their first treated patient, a 41-year-old man with multiply relapsed AML, who received an infusion of 1 × 109 CD33 CAR T cells (38% transduction efficiency) and experienced a partial response with marked reduction in marrow blasts prior to leukemia progression at 9 weeks from T-cell infusion. This patient also experienced clinically significant CRS with fever and elevated inflammatory cytokine levels. Importantly, the patient did not appear to have clinical or laboratory signs of sinusoidal obstruction syndrome/veno-occlusive disease, a potential on target/off tumor toxicity given CD33 expression on hepatic Kupffer cells. 89 This trial remains open for patient accrual.
Several additional phase I trials of CAR T cells for patients with AML are ongoing across the globe (Table 2). The MD Anderson Cancer Center (MDACC) is conducting a study of CD33-redirected CAR T cells and will allow pediatric patient participation if a safe and tolerable dose of T cells is first defined in adults with relapsed/refractory AML [ClinicalTrials.gov identifier: NCT03126864]. A separate Pediatric Bone Marrow Transplantation Consortium-sponsored CD33 CAR T-cell trial specifically for children, adolescents, and young adults with relapsed/refractory AML is also in development at the National Cancer Institute and Children’s Hospital of Philadelphia and anticipated to open in 2018.
Various centers in the United States [City of Hope Comprehensive Cancer Center, University of Pennsylvania (Penn) Abramson Cancer Center, Weill Cornell Medicine, MDACC] and in Beijing are conducting phase I trials of RNA-modified or lentivirally transduced CD123 CAR T cells. In the Penn trial [ClinicalTrials.gov identifier: NCT02623582], five adults with relapsed/refractory AML were treated with lymphodepleting chemotherapy followed by one to three doses of short-persisting RNA-electroporated CD123 CAR-modified autologous T cells administered every 48–72 h in a pilot trial to study manufacturing feasibility, safety, and potential myelotoxicity. 90 In this study, less than 60% of planned T-cell doses were able to be manufactured successfully, and all treated patients had leukemia progression within a month of cell infusion. T-cell persistence (as measured by flow cytometry and PCR) was expectedly undetectable given the very short half life of these biodegradable mRNA-transfected cells. However, all patients experienced fever or CRS necessitating tocilizumab administration for toxicity management. In addition, no obvious hematologic, neurologic, or vascular toxicity was observed given CD123 expression in normal tissues. 90 Given the lack of anti-leukemia efficacy, but also minimal on target/off tumor toxicity, this trial was terminated early with plans for development of a subsequent lentiviral CD123 CAR T-cell trial. The City of Hope group also recently reported early results from its phase I CD123 CAR T-cell trial [ClinicalTrials.gov identifier: NCT02159495]. 87 Six adults with relapsed AML after HSCT were treated with 50 × 106 (dose level 1, DL1) or 100 × 106 (DL2) lentivirally transduced CAR+ CD28/CD3ζ autologous T cells following lymphodepleting chemotherapy. Retreatment with a second T-cell infusion was permitted for eligible patients. One of two DL1 patients had transient morphologic remission and then responded to a second cell dose upon AML reprogression, ultimately achieving an MRD-level disease response. Two of four DL2 patients had complete responses (CRs) following CD123 CAR T-cell treatment and were able to undergo a second HSCT. The other two patients treated at DL2 had partial responses. Grade 2 or less CRS was observed in most patients, and no dose-limiting toxicity, including myeloablation, has been observed to date. A patient with a refractory blastic plasmacytoid dendritic cell neoplasm (BPDCN) treated with these CD123 CAR T cells also experienced a CR. 87 This trial is currently enrolling.
Additional phase I trials of CAR T cells for adults with relapsed/refractory AML include those targeting the transmembrane NKG2D C-type lectin-like receptor or its cell surface ligand (NKG2DL) at Dana-Farber Cancer Institute [ClinicalTrials.gov identifier: NCT02203825] or at several US and European sites [ClinicalTrials.gov identifier: NCT03018405].92–94 A phase I trial in Shenzhen, Guandong, China is testing the safety of CD38, CD56, CD117, or Muc-1 CAR T cells in children and adults with relapsed/refractory AML [ClinicalTrials.gov identifier: NCT03222674]. No data are yet publically available from these trials (Table 2).
Conclusion
The remarkable cure rates achieved in patients with multiply relapsed B-ALL treated with antigen-redirected CAR T cells has had transformative scientific and clinical impact and recently led to expedited FDA approval of two CD19 CAR T- cell products, tisagenlecleucel and axicabtagene ciloleucel. While similar successes are certainly equally desired for patients with AML treated with immunotherapy, issues of myeloid antigen selection for CAR targeting and the delicate balance of on target/on tumor and on target/off tumor toxicity remain significant unsolved challenges. Moderate to longer-term persistence of AML CAR T cells likely required for achievement of deep molecular remissions is potentially incompatible with maintenance of normal hematopoiesis. The sequela of CAR T-cell-induced myeloablation may be rescued with allogeneic HSCT, but steps must be taken to ensure that engineered T cells do not attack incoming donor cells and induce graft rejection.
Strategies to optimize AML CAR T-cell activity and reduce potential resistance are critically important for successful actualization of these new immunotherapies. In addition to the aforementioned bispecific antigen-targeting approaches to maximize AML specificity, use of genome-editing technologies, such as clustered regularly interspaced short palindromic repeats (CRISPR)/CRISPR associated protein 9 (Cas9) and transcription activator-like effector nucleases (TALEN), or checkpoint inhibitors may further fine-tune AML CAR T-cell precision, potency, and persistence.28,95–97 Combining CAR T cells with relevant kinase inhibitors may also decrease potential immunotherapeutic resistance leading to leukemia relapse. This concept has been recently successfully demonstrated in preclinical studies of CD19 CAR T cells and ibrutinib in ALL, FLT3 CAR T cells with the FLT3 inhibitor crenolanib in AML, and CD33 or CD19 CAR T cells with the PI3K inhibitor LY294002 in AML and ALL, respectively.59,70,98 Finally, successful engineering and treatment of patients with allogeneic donor-derived 99 or universal CAR T cells 62,88 without appreciable graft-versus-host or host-versus-graft reactivity may minimize typical delays often occurring between autologous T-cell pheresis to manufacture to infusion, as well as more readily increase therapy availability to a larger patient population in need.
As nascent bench-to-bedside translation of cellular immunotherapies for patients with relapsed/refractory AML is beginning to occur, biologic questions persist that may only be answerable with further clinical experience and careful monitoring of patients: (1) What are optimal AML-associated surface antigens (or antigen pairings) for targeting with CAR T cells and other immunotherapies? (2) Is there a therapeutic window for AML CAR T cells to maximize leukemia remission and minimize hematologic and non-hematologic toxicity? (3) Will hematopoietic stem cell transplantation be necessary to consolidate potential CAR T-cell-induced remissions or to rescue patients from on target/off tumor myeloablation? (4) Should AML CAR T cells be terminated via suicide switches or antibody depletion, particularly prior to HSCT, minimize allogeneic graft rejection? (5) Can HSCT donor-derived or ‘off the shelf’ allogeneic T cells successfully induce deep leukemia remission with minimal or tolerable graft-versus-host and host-versus-graft effects? (6) Will combining AML CAR T cells with checkpoint inhibitors or kinase inhibitors increase anti-leukemia efficacy or decrease immunotherapeutic resistance? (7) Can gene-editing strategies such as CRISPR/Cas9 and TALEN be used for further optimization of AML CAR T-cell specificity, persistence, toxicity, and availability to patients?
Footnotes
Acknowledgements
SKT wrote the manuscript.
Funding
This work was supported by a National Cancer Institute 1K08CA184418 career development award, the Rally Foundation for Childhood Cancer Research, the Gabrielle’s Angel Foundation for Cancer Research, and a St Baldrick’s Foundation/Stand Up to Cancer Pediatric Dream Team Translational Research Grant (SU2C-AACR-DT2727). Stand Up To Cancer is a program of the Entertainment Industry Foundation administered by the American Association for Cancer Research.
Conflict of interest statement
SKT has no relevant financial conflicts of interest.