
Abstract
Although cure rates for pediatric acute lymphoblastic leukemia (ALL) have now risen to more than 90%, subsets of patients with high-risk features continue to experience high rates of treatment failure and relapse. Recent work in minimal residual disease stratification and leukemia genomics have increased the ability to identify and classify these high-risk patients. In this review, we discuss this work to identify and classify patients with high-risk ALL. Novel therapeutics, which may have the potential to improve outcomes for these patients, are also discussed.
Incidence and outcomes
Acute lymphoblastic leukemia (ALL) is the most common malignancy in childhood. Annual incidence in the United States is approximately 30 in every million children under the age of 20.1,2 This results in approximately 3000 new cases of pediatric ALL annually. Incidence peaks during the first 2–5 years of life due to the expression of early or prenatally acquired somatic mutations in immature lymphoid cells, many of which inherited germline cancer predisposition polymorphisms or variants.3–5 Due to the success of sequential clinical trials, long-term survival for this disease now exceeds 90%.6–13 Much of this success has been driven by refinements in risk stratification, therapy intensification for higher risk groups, therapy de-intensification for lower risk groups, and more recently, novel, targeted therapies.
Early efforts to refine therapy aimed to utilize easily identified clinical characteristics to stratify patients by relapse risk. The National Cancer Institute (NCI) criteria separates patients into two risk groups based on the presenting age and white blood cell count: patients aged between 1 and less than 10 years who have a white blood cell count of less than 50 × 109/L are classified as standard-risk and those not meeting these criteria are classified as high-risk. 14 This classification best applies to B-cell ALL, with all patients with T-cell ALL requiring more intensive chemotherapy regardless of initial presenting age and leukocyte count. Despite the use of risk-directed therapy, patients with NCI standard- and NCI high-risk disease continue to have disparate outcomes, due mainly to a combination of somatic and germline genetic abnormalities.
Standard treatment for ALL includes combination chemotherapy using agents mostly developed in the 70s and 80s and lasts between 2 and 2.5 years. Although effective, acute toxic effects and long-term sequela are common.13,15–17 Recent protocols have further intensified therapy for very-high-risk groups while de-intensifying therapy for others in attempts to maximize cure while simultaneously minimizing acute and long-term toxicity. However, gains in outcomes for very-high-risk groups through intensification of conventional chemotherapy have reached their limit and reductions in relapse risk have been counterbalanced by increased toxicity, including death, in recent clinical trials.13,18 Thus, use of novel targeted therapies will be required to further improve cure rates while minimizing toxicity.
Therapy toxicity has been a particular challenge in adolescents who are universally categorized as NCI high-risk due to their older age. When treated with identical therapy on the NOPHO2008 protocol, the risk of toxic death from therapy was higher (5% versus 2%) than that of children <10 years. 12 Patients in this age group experience more thrombosis and hyperglycemia than younger children, 16 partly due to their delayed clearance of dexamethasone. 19 Adolescent patients treated on the AALL0232 protocol experienced a higher incidence of mucositis during interim maintenance than younger children, regardless of their assignment to Capizzi or high-dose methotrexate regimens. 17 Osteonecrosis risk is also significantly increased in this age group, particularly the most severe manifestations of osteonecrosis that require surgical intervention.20–22 Despite these toxicities, disease free survival (DFS) and overall survival (OS) are clearly improved for this population when treated with pediatric rather than adult regimens.23–25 Several excellent reviews of treatment in this population have previously been published and readers are referred to these for further discussion of issues specific to care for this high-risk population.23,26
Recent attempts at risk refinement, identification of high-risk subgroups of ALL, as well as novel agents now making their way into frontline therapy will be the focus of this review. While the definitions of high- and very-high-risk differ between treatment groups and protocols (Table 1), we will focus on features that appear to confer additional risk in multiple settings across different treatment regimens. Because of dramatic differences in therapy, risk stratification, and genetics between B-cell and T-cell ALL, the discussion herein will focus on B-cell ALL and the readers interested in T-cell ALL are referred to another recent review. 27
Risk stratification in recently completed trials.
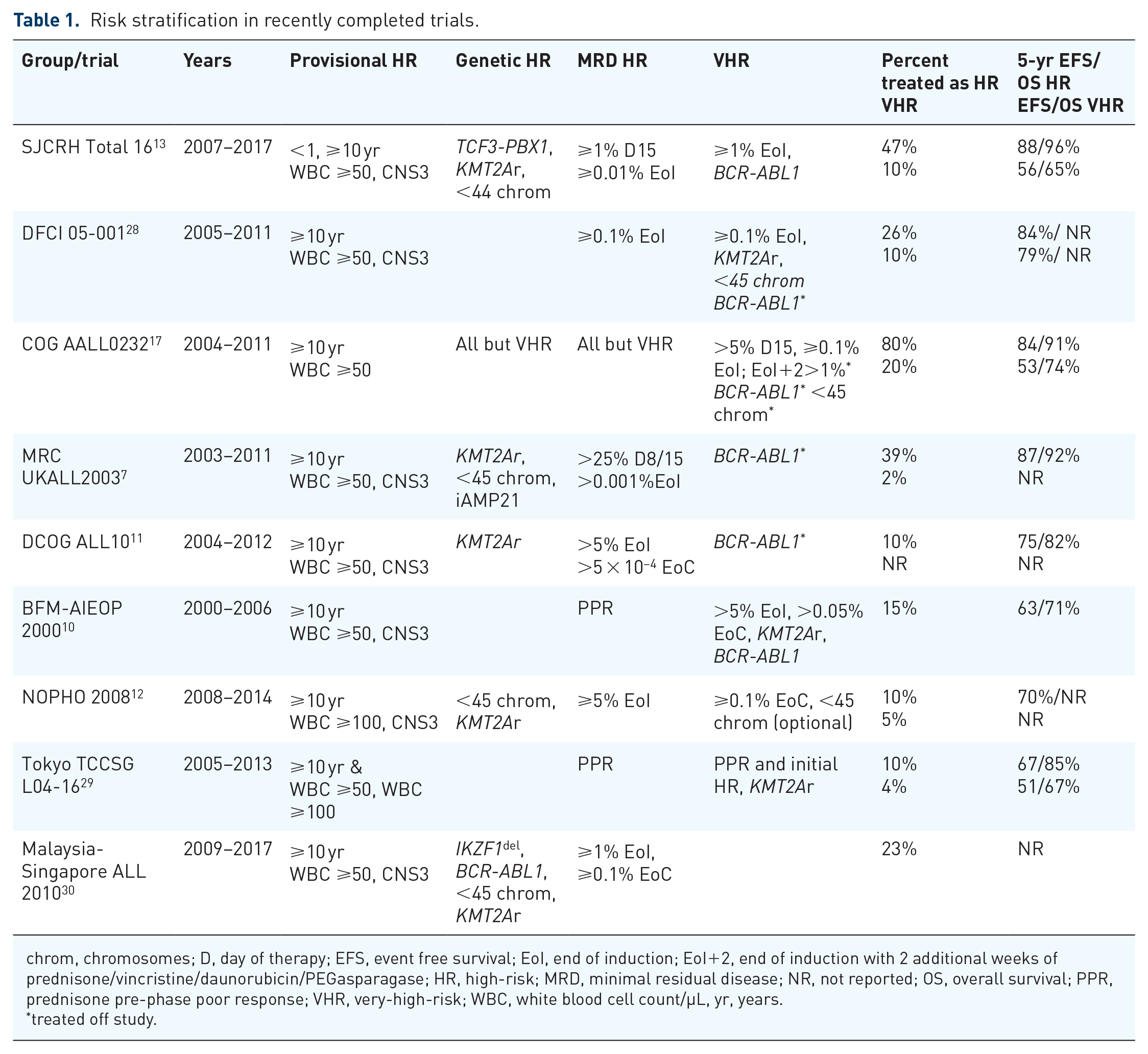
chrom, chromosomes; D, day of therapy; EFS, event free survival; EoI, end of induction; EoI+2, end of induction with 2 additional weeks of prednisone/vincristine/daunorubicin/PEGasparagase; HR, high-risk; MRD, minimal residual disease; NR, not reported; OS, overall survival; PPR, prednisone pre-phase poor response; VHR, very-high-risk; WBC, white blood cell count/µL, yr, years.
treated off study.
Minimal residual disease
Minimal residual disease (MRD) utilizes leukemia specific antigen or genotypic changes to identify sub-microscopic levels of persistent disease. Multi-color flow cytometry has routinely been able to identify leukemia at the level of 0.01% 31 and is now the preferred method used by most study groups because a reliable result can be obtained rapidly for treatment decisions. In comparison studies, MRD has consistently been more predictive of sustained remission and leukemia-free survival than morphologic remission.32–37 Next generation sequencing (NGS) assays are able to detect leukemia burdens at least a log lower than current technologies with flow cytometry, or immunoglobulin or T-cell receptor rearrangement by polymerase chain reaction (PCR).38–40 In a recent study, the NGS assay identified MRD at the conventional clinical cutpoint in more patients than flow cytometry, with discrepant patients having worse outcomes than other patients with negative MRD by flow cytometry. 41 Moreover, the NGS method can identify 20% of standard-risk B-ALL patients without any detectable level of MRD who can essentially be cured with current chemotherapy, with a 5-year event-free survival (EFS) of 98% and OS of 100%. 41 Whether this newer assay can be incorporated routinely into frontline trials will require improved technology, allowing for results to be readily available with local rather than central testing.
Intensification of therapy based on MRD
St. Jude Children’s Research Hospital
St. Jude Children’s Research Hospital has used MRD determined by flow cytometry prospectively for risk-directed treatment since 2000. MRD is determined after 2 weeks of remission induction therapy and at the end of 6-week remission induction. This information, combined with age, presenting white blood cell count, and genetics, has stratified 43% of patients to be treated as low-risk [age ⩾1 but <10 years and an initial white blood cell (WBC) count of <50 × 109/L, presence of either a DNA index ⩾1.16 or ETV6-RUNX1 fusion without adverse genetic or clinical feature [central nervous system (CNS) or testicular leukemia], or slow response to therapy (MRD < 1% after 2 weeks of therapy and <0.01% at the end of induction)] and the remainder as either standard-risk (47%) or high-risk (10%; Table 1).13,42
Patients with provisional low-risk disease (based on presenting clinical and genetic features) and less than 1% MRD after 2 weeks of therapy and less than 0.01% MRD at the end of 6-weeks of induction therapy had excellent 5-year EFS of greater than 95%.13,42 Among provisional low-risk patients, those with MRD of less than 1% after 2 weeks of induction therapy but who had MRD between 0.01% and 0.99% at the end of induction also had an excellent EFS (100% at 10 years) with intensified post-remission therapy. 43 By contrast, provisional low-risk patients with more than 1% MRD after 2 weeks of therapy had inferior 10-year EFS, with only 69% long-term EFS. However, these patients could be further risk stratified based on their end of induction MRD, with the patients without detectable MRD (<0.01%) having an EFS of 89%, those with low-level MRD have an EFS of 67%, and those with MRD of greater than or equal to 1% having an EFS of only 25%. 43 Hence, early MRD result is useful to identify provisional low-risk patients who are highly curable.
Similar trends were observed among patients with provisional standard-risk disease (those meeting NCI high-risk criteria or with adverse biological features). 43 In this population, greater differences were observed in patients based on their 2-week bone marrow assessment: EFS of ~83% in those with MRD of less than 1% compared with 65% in those with higher levels of MRD at the 2-week time point. The trend towards improved outcomes in patients without detectable MRD (<0.01%) at the end of induction despite detectable MRD after 2 weeks of therapy was maintained in this group. Importantly, only 2 patients of 11 with detectable (>0.01%) but declining MRD between the end of induction and the start of reinduction/delayed intensification experienced relapse, suggesting that ongoing intensified chemotherapy may be sufficient in such patients as long as MRD negativity is obtained prior to this phase of therapy. Notably, MRD and genetic features both have independent prognostic significance, and should be used in concert for risk-directed therapy. Patients with hyperdiploid ALL or an ETV6-RUNX1 fusion, two groups with historically favorable outcomes together comprising ~40% of pediatric ALL, had excellent outcomes, particularly in the presence of negative MRD. In contrast, patients with adverse genetic features have higher risks of relapse despite MRD negativity. 44
Children’s Oncology Group
The Children’s Oncology Group (COG) has historically used a flow cytometric assay to evaluate MRD. In the AALL0232 trial for NCI high-risk patients, patients with an MRD of greater than 0.1% at the end of induction received intensified therapy utilizing two delayed intensification and interim maintenance cycles. 17 In the context of this MRD intensified therapy, end of induction MRD levels remained prognostic, with patients with rapid early response (M1 on day 15 and MRD less than 0.1% at end of induction) having 5-year EFS of 83.9% versus 53.3% in those with slow response (MRD above 0.1% at end of induction or an M2/M3 marrow on day 15 of induction). Interestingly, patients who received intensified therapy due to slow early response (as defined above) had similar 5-year EFS but with different relapse kinetics than patients whose therapy was not intensified but who had MRD detectable at less than 0.1% at the end of induction. 45 In patients with intensified therapy, 5-year EFS was 63% with 23% of relapses occurring in the first 2 years of remission; in contrast, 5-year EFS was 74% in patients with detectable MRD below 0.1% (who did not receive intensified therapy), with 52% of relapses occurring in the first 2 years of remission. These data suggest that intensification of therapy for these patients may have delayed but not prevented relapse. As a result of these and other data, patients with detectable MRD of greater than 0.01% have received intensified therapy on subsequent protocols. Notably, clearance of MRD (to <0.01%) by the end of consolidation was associated with a relatively favorable 5-year EFS of 79% compared with 39% in patients with MRD at this threshold after consolidation. In current protocols, MRD is obtained at the end of consolidation for patients with end of induction MRD to further risk stratify for therapy. Patients with clearance of MRD remain on protocol receiving chemotherapy while those with persistent disease are being directed to cellular therapy, with a recommendation to receive chimeric antigen receptor T-cell (CART) therapy as part of a clinical trial (NCT03876769).
Medical Research Council United Kingdom
Initial clinical risk stratification in UKALL2003 was refined by end-of-induction MRD assessment in a subset of patients. Specifically, patients with a rapid early bone marrow response (<25% blasts on day 8 for NCI high-risk or those with adverse genetics, and on day 15 for NCI standard-risk) were included in a stratified randomization based on MRD determined by PCR of leukemia specific immunoglobulin or T-cell receptor rearrangements. 7 Patients with MRD <0.01% at end of induction and without detectable leukemic rearrangement (threshold of quantification 0.01%) before interim maintenance were deemed low-risk, those with detectable MRD at <0.01% before interim maintenance were categorized as intermediate-risk, and those with MRD at ⩾0.01% at the end of induction were categorized as high-risk. Intensified therapy in this high-risk group, which utilized escalating-dose methotrexate combined with pegylated (PEG)-asparaginase (Capizzi regimen) for interim maintenance and additional vincristine and asparaginase doses in delayed intensification, improved survival compared with standard therapy (5-year EFS 89.6% versus 82.8%, p = 0.04). Although which of these therapeutic changes is responsible for the improved outcomes is unclear, data from Dana Farber suggests that additional weeks of PEG-asparaginase likely contributed to this improved outcome. 46
As in the St. Jude data, there was an interaction between the genetic/clinical risk group and MRD-associated relapse risk in UKALL2003. 47 Elevated end-of-induction MRD was associated with an increased risk of relapse in all groups identified. However, the hazard associated with MRD was greater in those with pre-defined higher-risk clinical or genetic features. For example, patients with favorable genetics had a 5-year relapse risk of 3% if MRD was undetectable, 4% if MRD was detectable at <0.01%, and 6% if MRD was 0.01–<0.1% (although this last group received intensified high-risk therapy). In contrast, the relapse risk in the adverse genetics group was 12%, 43%, and 10%, respectively, although the number of patients in each group was small (25 or less), decreasing the precision of these estimates.
Summary and future directions
Together, these data highlight two key findings from recent studies: the benefit of intensifying therapy for patients with detectable MRD at ⩾0.01% at the end of 3- or 4-drug induction therapy, and the importance of combining traditional risk factors, especially genetics, with MRD to determine the true relapse risk after achieving first remission. Risk stratification combining genetic and MRD risk data from recent group trials are summarized in Table 1.
The use of NGS assays to identify MRD is being studied across multiple groups. Such assays can typically detect dominant sequences in >90% of patients. 48 Data from the Children’s Oncology Group suggest that patients who are flow cytometry-negative but have detectable MRD by NGS have inferior outcomes to those without detectable NGS MRD. These differences appear to be largely driven by patients with a detectable disease by NGS at greater than 0.01% who were considered negative by flow cytometry. Additional differences are seen in the NCI standard-risk population, where patients who were NGS negative have excellent EFS (98%) versus 92.7% in those with detectable NGS-MRD. 41 The implications of persistent MRD will also require continued validation as novel therapies are introduced, including those targeting leukemic antigens (i.e. CD19 and CD22) which may abrogate the adverse prognosis associated with MRD.
Genetic subgroups altering risk
Philadelphia chromosome positive ALL
The adverse effect of the t(9;22)(q34;q11.2)/BCR-ABL1 fusion in patients with ALL has long been recognized. Prior to the advent of tyrosine kinase inhibitor (TKI) therapy, outcomes were poor with long-term EFS and OS of 31% and 44%, respectively, despite the frequent use of allogeneic stem cell transplantation in first remission. 49 Transient single-agent activity was shown with imatinib, a first-generation ABL tyrosine kinase inhibitor; incorporation of imatinib into intensive chemotherapeutic backbones occurred in the early 2000’s. These combinations were well tolerated without increased toxicity. The 3-year EFS in the COG AALL0031 study for children obtaining a remission after induction therapy was 80% in the final dose cohort, superior to patients treated in the pre-TKI era. 50 These results appeared durable with longer follow-up, with 5-year DFS of 70%. 51 The effectiveness of imatinib also abrogated the advantage of transplantation in first complete remission (CR).
Results from the European intergroup EsPhALL provided further evidence of the efficacy of imatinib in pediatric Ph+ ALL. 52 Despite intermittent dosing in patients randomized to receive it, imatinib improved outcomes in patients with a good prednisone pre-phase response (4-year DFS 72.9% versus 61.7% in those assigned chemotherapy alone). In patients with a poor prednisone response, all of whom received imatinib, the 4-year EFS was 53.3% compared with 10% in a historical control.
The early introduction of TKI therapy also appears to benefit patients by improving early response to therapy. The AALL0622 study utilized dasatinib, a second-generation ABL TKI with broader activity against BCR-ABL mutations and additional activity against SRC kinases. 53 Unlike in prior trials which initiated imatinib after induction, dasatinib therapy at 60 mg/m2 per day was begun on day 15. This early introduction of TKI increased both the end of induction CR rate (98% versus 89% in AALL0331) and the rate of MRD-negative remissions (59% versus 25%). 54 Despite these improvements in early response, long-term outcomes were similar to imatinib based therapy, with a 5-year OS and EFS of 86% and 60%, respectively. A recent randomized study suggested that dasatinib given at 80 mg/m2 is superior than imatinib to increase EFS and decrease isolated CNS relapse without the need of prophylactic cranial irradiation or allogeneic transplantation. 55
Importantly, Ph+ ALL has demonstrated the effectiveness of targeted therapy when combined with conventional chemotherapy and remains the exemplar of precision medicine in acute hematological malignancies. The tolerability of these regimens has also allowed the expanded use of TKI to encompass patients with Ph-like ALL (discussed further below). Studies in adults have demonstrated the short-term effectiveness of novel agents in Ph+ ALL, including the third generation TKI ponatinib, antigen directed therapies such as blinatumomab and inotuzumab, and novel agents such as venetoclax.56,57 Further follow-up of these agents in adults and evaluations in pediatric populations may allow further improvement in outcomes for these very-high-risk patients.
Philadelphia-like ALL
Philadelphia chromosome-like ALL was first identified as a group of B-cell ALL with a gene expression pattern similar to that of BCR-ABL1 positive ALL but lacking the classic fusion sequence. 58 These leukemias comprised 12% of pediatric ALL compared with Ph+ ALL which comprises 3–4% of pediatric ALL. 59 These leukemias were initially identified as having frequent deletions of IKZF1, similar to Ph+ ALL, as well as a poor prognosis similar to Ph+ ALL with a 5-year EFS of 60%. Subsequent work demonstrated the frequent activation of tyrosine kinases, including ABL1 and ABL2 via novel fusions, PDGFRB, CSF1R, CRLF2, JAK2, and EPOR. 60 Preclinical data demonstrated the sensitivity of these kinase-activated leukemias to kinase inhibition either via dasatinib or ruxolitinib. Although retrospective data suggests that MRD-guided therapy could yield treatment outcome in patients with Ph-like ALL comparable with that of other B-ALL patients, 15% of Ph-like ALL were transplanted due to MRD ⩾1% at the end of remission induction. 60 Thus, it is important to identify Ph-like ALL patients with tyrosine kinase activation for targeted therapy to spare them from intensive chemotherapy or transplantation.
Indeed, early studies showed that patients with Ph-like ALL and targetable ABL class fusions had poor response to conventional chemotherapy but would respond to ABL-class TKI in combination with standard therapies. 61 These efforts have now been expanded to frontline clinical trials. Similar success has yet to be reported for ruxolitinib in cases with JAK activation and results of multiple ongoing clinical studies are eagerly awaited.
KMT2A rearrangements
Rearrangement of the KMT2A (formerly MLL) locus at chromosome 11q23 is associated with an inferior prognosis.62–64 While these translocations are genetically diverse, they all appear to have inferior prognosis to leukemias not carrying these rearrangements. The prognosis appears to be particularly poor in children less than 1 year of age. In these infants, 70% carry somatic KMT2A rearrangements. 65 Infants with a KMT2A rearrangement have a DFS of less than 40%.65,66 The prognosis is particularly poor in extreme young age (defined by different groups as less than either 6 or 3 months), in patients with high white blood cell counts, and in those with CNS involvement at diagnosis.
Because of their unique biology as well as their unique sensitivity to chemotherapy induced complications, infants are typically treated on stand-alone protocols. Non-infants carrying KMT2A rearrangements are typically treated on the high-risk or very-high-risk arms of pediatric protocols.
Unlike in Ph+ ALL with BCR-ABL fusion, targeted therapies that are effective in infants have not yet been identified in clinical practice. Clinical trials have targeted dysregulation of epigenetic modifiers as well as overexpression of FLT3. Although final results of the studies have not been published, preliminary preclinical data suggest that menin inhibitors as well as venetoclax may have activity in children with KMT2A rearrangements.67–70 Notably, patients with this leukemia appear to fare poorly with antigen directed therapy due to the propensity of KMT2A rearranged leukemias to undergo lineage shift or antigen loss when CD19 or CD22 directed therapies are used.71–73 This has been observed with both blinatumomab as well as CD19 directed chimeric antigen directed T-cell therapy.
Hypodiploid
Hypodiploid ALL, defined as having fewer than 44 chromosomes, is associated with an adverse prognosis. In retrospective data gathered between 1986 and 1999, 72 patients with hypodiploid ALL had a 5-year EFS of less than 45%. 63 Hypodiploid ALL can be subdivided into biologically distinct categories based on modal chromosome number: near haploid with 23–29 chromosomes, low hypodiploid with 30–39 chromosomes, and high hypodiploid with 40–43 chromosomes. Patients with low hypodiploid ALL have mutations in TP53 in >90% of cases, with half of these being constitutional (i.e. having Li Fraumeni syndrome). 74 In contrast, patients with near haploid ALL have a high frequency of RAS family and receptor tyrosine kinase mutations (>70%).
These biologically distinct subsets have different clinical courses. High hypodiploidy patients have a 5-year EFS of 74% compared with 48% in other hypodiploid cases. 75 MRD remained highly prognostic in hypodiploid cases, with patients achieving MRD-negative remission (<0.01%) having a 5-year EFS of 75% compared with 49% in those with detectable MRD. While transplantation did not improve survival in either MRD-negative or MRD-positive hypodiploid ALL, treatment on an MRD-directed chemotherapeutic protocol was associated with improved survival, presumably due to intensification of therapy for appropriate patients. In addition to the increased risk of treatment failure and need for close MRD monitoring in patients with hypodiploid ALL, the presence of germline/constitutional TP53 mutations poses other hazards for patients. The risk of second malignancy in patients with germline TP53 mutations was 35-fold higher than other patients in a recent cohort of high-risk ALL. 76 These data suggest that avoidance of therapy known to increase the risk of second neoplasms (i.e. epipodophyllotoxins and radiation) should be emphasized in these patients. Novel agents may therefore be needed. To this end, preclinical data has suggested that venetoclax may be active against hypodiploid ALL. 77 Future trials evaluating this combination should be considered.
Intrachromosomal amplification of chromosome 21 (iAMP21)
Amplification of a portion of the long arm of chromosome 21 including the RUNX1 locus were first noted as recurrent lesions in 2003.78,79 While variable in cytogenetic appearance, the defining feature of this chromosomal abnormality is a focal amplification of the region including the RUNX1 locus, resulting in at least 5 copies of RUNX1 on a single chromosome 21. Comprising approximately 2% of pediatric ALL, iAMP21 patients tend to be older and are roughly evenly divided between NCI standard- and high-risk.63,79 Early reports suggested that this subtype was associated with a poor prognosis, with a 3-year EFS of approximately 25%. 80 Relatively high salvage rates for these patients suggested that intensification of therapy could improve outcomes. Based on these and other data, the MRC began treating all patients with iAMP21 as high-risk, regardless of other characteristics. 81 This resulted in an increased in EFS from 29% to 78% and a reduction in relapse risk from 70% to 16%. Contemporaneous COG protocols demonstrated an inferior outcome for iAMP21 patients treated on NCI-standard risk but not NCI-high risk protocols. 82 This finding was replicated in a retrospective international intergroup study. 83 As a result of these data, most trials now classify iAMP21 patients as high-risk and treat them with intensified therapy regardless of other clinical characteristics.
MEF2D rearrangements
NGS has led to the identification of several novel rearrangements that are not evident on conventional genetic analysis, including MEF2D rearrangement.84–86 This genotype occurred in 2–5.5% of B-ALL with an older age at diagnosis (median 12 years), a poor OS (33–70%), and an association with upregulation of pre-B-cell receptor signaling molecules but downregulation of JAK/STAT signaling pathway. Preclinical studies suggested that this genotype is sensitive to histone deacetylase inhibitors such as panobinostat and vorinostat, but clinical studies are lacking.
IKZF1 deletion
Deletion of the IKZF1 gene encoding the B-cell transcriptional factor Ikaros was first noted in Ph+ ALL. 87 Subsequent evaluation of cohorts of NCI high-risk ALL demonstrated IKZF1 deletions in ~28% of patients, a rate higher than seen in a second cohort used to validate clinical outcomes. 59 Patients in that cohort treated on high-risk protocols without MRD-based intensification had a 5-year EFS of less than 50%. Subsequent evaluations of NCI standard- and high-risk cohorts suggested that IKZF1 deletion was associated with a 2.2-fold increase in relapse risk in high-risk patients but a non-significant 1.5-fold increased risk in standard-risk patients. 88 Other analyses have shown that IKZF1 deletions carry an adverse prognosis across all risk groups. 89 Recent work has suggested the IKZF1 deletion’s adverse prognosis is worsened by co-occurring deletions in any of: CDKN2A, CDKN2B (homozygous deletions only), PAX5, or PAR1, all in the absence of ERG deletion. 90 This group, termed IKZF1plus, comprises ~6% of pediatric ALL and is associated with a more adverse prognosis than IKZF1 deletion alone, particularly in patients with MRD detectable above 0.01% at the end of either induction or consolidation.
Intensification of therapy may partially ameliorate the adverse prognosis of patients with IKZF1 deletions. Patients with deletions benefitted from maintenance dexamethasone/vincristine pulses in the EORTC 58591 study. 91 Separately, in the Malaysia-Singapore 2010 study, any patients identified to have IKZF1 deletion were assigned to the next higher level of risk-directed therapy. 30 Hence, all 50 patients with IKZF1 deletion received intensified treatment in the intermediate-risk (n = 25) or high-risk arms (n = 25), and none received less intensive treatment in the standard-risk arm. The 50 patients with IKZF1 deletion treated in this study had significantly lower cumulative risk of relapse (13.5% versus 30.4%) and improved 5-year OS rate (91.6% versus 69.6%) as compared with the 59 historical controls who were retrospectively identified to have IKZF1 deletion and treated with standard-risk (n = 13), intermediate-risk (n = 20), or high-risk arms (n = 20) of the preceding MS 2003 study. Notably, none of the 13 standard-risk patients with IKZF1 deletion who had negative MRD level (⩽0.01%) at the end of induction relapsed after treatment in the standard-risk arm of MS 2003 study. Thus, genetic data should be used in concert with MRD assessment for risk-directed therapy.
New agents
The advent of antigen directed therapy for pediatric ALL has significantly altered the therapeutic landscape. Blinatumomab, a bispecific CD-19/CD-3 antibody, brings T-cells in close proximity to CD-19 expressing lymphoblasts and triggers T-cell directed cytotoxicity. Early studies of adults with relapsed or refractory leukemia demonstrated that blinatumomab was able to achieve higher rates of response (44% versus 25%, p < 0.001) and longer median OS (7.7 versus 4 months, p = 0.01) than standard chemotherapy. 92 Blinatumomab appeared to be even more effective in adults with MRD-positive remissions, with 78% of 113 patients achieving an MRD-negative remission. 93 Based on these encouraging adult data, a phase I/II study of blinatumomab in pediatrics was undertaken. 94 Results were similar to the adult study, with 39% of these relapsed/refractory patients treated at the phase II dose achieving remission. Although the data are more limited, blinatumomab also appears to be effective in eliminating MRD prior to transplant in patients in remission. A single cycle of blinatumomab was effective in eliminating MRD in 14/15 patients with residual MRD after consolidation therapy in a retrospective cohort. 95 The therapy was well tolerated and the 1-year survival was 93%. A recent Children’s Oncology Group’s study showed that the substitution of blinatumomab compared with conventional intensive consolidation therapy significantly improved the 2-year DFS of patients with relapsed or refractory ALL (59.3% ± 5.4% versus 41.0% ± 6.2%). 96
Inotuzumab ozogamicin, an antibody-drug conjugate with calicheamicin, targets CD22, an antigen expressed on more than 90% of B-cell ALL cases.97,98 Although the mechanisms of action of blinatumomab and inotuzumab are different, results from early inotuzumab studies also showed benefit over conventional chemotherapy. Response rates of 57–68% were seen in adult phase I/II studies,99–101 and phase III studies demonstrated higher remission rates (81% versus 29%, p < 0.001) and longer median survival (7.7 versus 6.7 months, p = 0.04) than standard chemotherapy in adults with relapsed ALL. 102 Although prospective data in pediatrics are currently lacking, a retrospective evaluation of inotuzumab compassionate use in 51 relapsed/refractory pediatric patients demonstrated a 67% response rate, including 48% of patients (71% of responders) who were MRD-negative. 103 Inotuzumab has also been combined with low-intensity chemotherapy in adults. In this relapsed population, inotuzumab combined with mini-hyper CVD resulted in a 75% overall response rate and 43% 1-year OS rate, both superior to historical control therapy with inotuzumab alone. 104
Antigen-directed therapy with CART has shown even more promising results. These genetically modified T-cells express an antibody against either CD-19 or CD-22 linked to a transmembrane domain and both a T-cell activation signal as well as a costimulatory domain. Early reports of tisagenlecleucel, a CD-19 targeted CART with a 4-1BB costimulatory domain, suggest a high response rate, with remission in 90% and a 6-month EFS of 67% of the first 30 patients. 105 In a multicenter phase II study, 81% of 75 infused patients achieved a remission. 106 Notably, 32 screened patients did not receive product, including 17 who were enrolled but either failed product production (7), became too ill for therapy (3), or died prior to infusion (7). The 12-month EFS was 59% among patients achieving a complete response, implying that 36 patients achieved that mark. CD-19 targeted CART utilizing CD-28 costimulation also have shown activity in relapsed pediatric ALL, with a 70% response rate in 20 treated patients during the phase I study; however, the median follow-up duration was only 10 months with an OS of 51.6% at 9.7 months and beyond. 107 Failures in all cohorts have involved both CD-19 positive relapses and CD-19 negative “antigen escape” relapses. The factors which identify patients most likely to receive both short- and long-term benefit remain under investigation.108,109 Early reports suggest that lower-burden disease, fludarabine based lymphodepletion, and pre-infusion bone marrow function (as measured by platelet count) may influence durability of response. The development of CD-22 directed CART, and reports of their short-term efficacy, suggest future therapeutic options for patients with CD-19 loss following CD-19 directed therapies. 110 The apparent propensity of KMT2A rearranged leukemias to undergo lineage switch while under antigen-directed therapy suggests that careful monitoring for this and potentially alternative therapies may be needed for this subtype of ALL.71–73 Identifying this phenomenon and reporting other subtypes capable of such evasion strategies will be critical as these therapies are used more widely, as other fusion protein-driven ALL have also demonstrated this capability. 111
In the absence of antigen directed therapy, there is significant excitement around the development of targeted small molecule inhibitors against BCL-2 family members. The only currently US Food and Drug Administration-approved agent in this class is the BCL-2 inhibitor venetoclax. This agent has shown exciting preclinical activity against hypodiploid ALL, 77 KMT2A rearranged ALL,69,112,113 Ph+ ALL,114,115 and the highly chemotherapy resistant (and rare) TCF3-HLF fusion ALL.113,116 Clinical trials are currently underway evaluating the safety and efficacy of venetoclax in combination with a variety of classical chemotherapy agents, including pediatric-inspired steroid/vincristine/asparaginase combinations and mini-hyper CVD. Preclinical data suggest that resistance to this agent may be driven by overexpression of BCL-2 family members MCL-1 and BCL-xL.114,117,118 To this end, a clinical trial is currently underway evaluating the combination of venetoclax and navitoclax (a combination BCL-2/BCL-xL/BCL-w inhibitor) in children and adults with relapsed/refractory ALL.
Future directions
The past decade has resulted in the identification of novel subtypes of B-cell ALL with important differences in outcomes. At the same time, data on the implications of these lesions in the context of MRD-directed therapy have enabled determination of which lesions result in an adverse prognosis, even in the context of such response-adapted therapy. Ongoing studies are testing the role of targeted or intensified therapies for these subsets of ALL which have an adverse prognosis despite prior therapy changes and intensification. Risk stratification of these studies are shown in Table 2.
Risk stratification in select current trials.

chrom, chromosomes; D, day of therapy; EFS, event free survival; EoI, end of induction; EoI+2, end of induction with 2 additional weeks of prednisone/vincristine/daunorubicin/PEGasparagase; HR, high-risk; MRD, minimal residual disease; OS, overall survival; PPR: prednisone pre-phase poor response; NR, not reported; VHR, very-high-risk; WBC, white blood cell count/µL; yr, years.
treated off study.
While every effort has been made to reflect the complexities of protocol design and risk stratification in the above tables, subtle differences may not be reflected here (e.g. additional modifications to risk based on transplant donor availability, the presence of other modifying genetic factors, or additional risk groups which have been compressed into the high-risk category for presentation simplicity). The authors thank study principal investigators for their review of these tables.
Future studies may also adjust therapy based on individual responsiveness to traditional chemotherapy or novel agents. In vitro response to chemotherapy (e.g. as assessed by methyl-thiazol-tetrazolium assay) has previously been correlated to treatment response in large clinical cohorts.119–121 While these sensitivity profiles appear to predict patients at high-risk of poor treatment response, these results are largely congruent with MRD analysis. 122 However, when combined with genetic information, these profiles can predict decreased efficacy of chemotherapy, including antimetabolite therapy used when detectable disease is no longer available to measure treatment response. 123 It is hoped that such measures or similar pharmacotyping assays evaluating response to novel therapies including immunotherapy, immune-drug conjugates, and kinase inhibitors can be used to guide future therapy to further improve outcomes for patients with high-risk ALL.
Critical studies are needed in the coming years to identify the optimal way to combine novel therapies or integrate them into existing regimens to build on the successes of prior protocol optimizations. Simultaneously, ongoing work will be needed to identify which genetically defined subsets of leukemia will benefit most from the various novel interventions, and what the long-term toxicities are of these novel interventions. Given the expanded genetic diversity of high-risk leukemia which has been recently described, collaboration will be critical in enabling these future advances.
While every effort has been made to reflect the complexities of protocol design and risk stratification in the above tables, subtle differences may not be reflected here (e.g. additional modifications to risk based on transplant donor availability, the presence of other modifying genetic factors, or additional risk groups which have been compressed into the high-risk category for presentation simplicity). The authors thank study principal investigators for their review of these tables.