
Abstract
Multiple myeloma (MM) has a worldwide incidence of 1–5/100,000/year. Outcomes have improved significantly in recent years following incorporation of immunomodulatory drugs and proteasome inhibitors into standard-of-care regimes. MM is profoundly immunosuppressive, enabling immune evasion, proliferation and disease progression. The role of the immune system in MM is becoming increasingly characterized and understood, and numerous therapies are under development or in routine clinical use targeting these elements of MM pathogenesis. In this review we discuss the immunosuppressive effects of MM, then the therapies targeting these defects. Specifically, we review the monoclonal and bispecific antibodies, alongside adoptive cellular therapies currently under investigation.
Introduction
Multiple myeloma (MM) is a malignant neoplasm of clonal plasma cells, preceded in nearly all cases by an asymptomatic premalignant condition called monoclonal gammopathy of uncertain significance (MGUS). Incidence of MM is approximately 4/100,000/year, and MGUS occurs in 3–4% of individuals over 50 years of age, with an average rate of progression to MM of 0.5–1%/year. 1 Patient outcomes have improved significantly over recent years. Kumar and colleagues showed a 50% improvement in overall survival in patients diagnosed after 1997 compared with those diagnosed before, with a clear role played by the advent of immunomodulatory drugs and proteasome inhibitors. 2 Outcomes have likely improved further, and numerous additional therapeutic agents are now available.
The role of the immune system in myeloma is becoming increasingly understood. Therapies targeting the immunological components of myeloma genesis offer a means of continuing to improve disease outcomes while avoiding or further reducing dependence on traditional chemotherapeutics. Furthermore, evidence suggests that immune surveillance may play a critical role in the prevention of disease progression, therefore early use of immune-focused therapies may have the potential to halt progression altogether.
In this review, we discuss the current understanding of the effects on the immune system seen in MM and its premalignant stages, how these affect disease progression, and the therapies available or under development.
Dysfunctional immunity in myeloma
Within the bone marrow (BM), a complex interplay of interactions occurs between malignant plasma cells and normal BM components. In this section, we will describe myeloma’s immunosuppressive effects on various cell types and subsets, and how these facilitate immune evasion. These are summarized in Figure 1.

The major components of impaired immunity in myeloma.
T-cells
T-cells form a crucial part of the adaptive immune system. They express unique antigen-specific T-cell receptors (TCRs), however they rely on antigen presentation by major histocompatibility complex (MHC) class I (for CD8+ cells) or class II (CD4+ cells) molecules for activation.
Allogeneic stem cell transplantation (allo SCT) provided the initial evidence of the role of T-cells in myeloma. Graft-versus-host disease (GvHD), especially chronic disease, is associated with improved survival post-allo SCT, 3 with a 5-year overall survival (OS) of 78.8% for those with chronic GvHD reported, compared with 42.6% without. The anti-tumour properties of donor lymphocyte infusions (DLIs) also correlate with the induction of GvHD and the quantity of lymphocyte dose. 4
Quantitative and qualitative T-cell changes are seen at varying stages of myeloma development. The peripheral blood CD4+/CD8+ T-cell ratio is reduced in MGUS and MM, and progresses with disease progression and increasing tumour burden, which have been shown to be independently associated with poor prognosis. 5 Th1 cells are increased in both BM and peripheral blood compared with Th2 cells, and Th17 cells, a proinflammatory subset, are particularly prominent in BM samples. 6 Tumour-specific clonal T-cell expansions may develop, more often with CD8+ than CD4+. Michalek and colleagues 7 stimulated the development of tumour-specific T-cell clones in patients using dendritic cells (DCs) loaded with myeloma cell apoptotic bodies. These T-cells demonstrated cytotoxic effects against autologous tumour cells in vitro. Clonal CD8+ T-cell expansions are seen more frequently in patients with low tumour burdens, and T-cell populations from the BM of MGUS patients respond to autologous malignant cell exposure by vigorous cytokine production, which is not replicated using T-cells from myeloma patients, suggesting impaired functionality with disease progression. 8 These studies cannot determine whether the impaired T-cell repertoire predisposes to disease progression or is caused by it, however experimental data suggest that immune surveillance may be involved in the prevention of progression. For example, T-cells specific for SOX-2, a gene critical for embryonic renewal of stem cells, were identified in around 25% of MGUS patients but absent in myeloma patients. Immunity to SOX-2 prevented the in vitro clonal growth of MGUS cells, suggesting that the loss of anti-SOX-2 immunity may facilitate the clonal escape of MGUS cells and disease progression. 9
T-cell dysfunction in myeloma is multifactorial. DCs, the central antigen-presenting cells (APCs), are impaired in MM. MM cells may induce T-cell anergy by presenting tumour-specific antigens without co-receptor expression. Brown and colleagues showed reduced expression of the B7-1 (CD80) costimulatory molecule on MM cells alongside downregulation of its counter receptor molecule CD28 on expanded T-cell clones, leading to T-cell anergy. 10 These tumour cells still expressed CD86 (B7-2) which interacts with cytotoxic T-lymphocyte associated antigen-4 (CTLA-4), noted to be upregulated in the T-cells. CTL4 binding dampens effector T-cell activation and regulates immune homeostasis. Interactions between programme cell death receptor-1 (PD-1) and its ligand (PD-L1) are another mechanism of immune suppression. PD-L1 is expressed by various nonlymphoid cells and tumour cells. PD-1/PD-L1 binding suppresses the activation and proliferation of autoreactive T-cells, inducing T-cell exhaustion, reduced cytokine production and impaired cell lysis. PD-L1 also binds to B7-1, mediating T-cell inhibition. 11 Increased levels of PD-L1 in myeloma cells alongside T-cell exhaustion has been demonstrated, and PD-L1 blockade in mice was shown to improve survival post-stem cell transplant and whole-cell vaccination. 12
TIGIT (T-cell immunoglobulin and immunoreceptor tyrosine-based inhibitory motif domain) is another inhibitory immune receptor expressed on T-cells and natural killer (NK) cells. Increased TIGIT expression on T-cells has been noted in patients with MM during disease progression. These T-cells exhibited a dysfunctional phenotype and demonstrated impaired proliferation and cytokine production. Addition of a monoclonal antibody against TIGIT led to improved T-cell function and suppressed MM development. 13
Studies focused on specific T-cell subsets have provided further information. Regulatory T-cells (Tregs) are immunosuppressive and required for normal immune homeostasis. CD4(+)CD25(+/high)FoxP3(+) Tregs are elevated in the peripheral blood of myeloma patients, with levels correlating with disease burden, and also seen in MGUS, suggesting a possible role in early myeloma genesis. Furthermore, myeloma cells have been shown to induce the formation of immunosuppressive Tregs in vitro. 14 NK T-cells express both TCRs and NK cell surface markers, recognizing antigens via CD1d molecules. Invariant NK T-cells (iNKTs) involved in tumour immunosurveillance, have been shown to be functionally impaired in myeloma patients with a reduced ability to produce interferon gamma (IFN-γ), possibly relating to the loss of CD1d expression by MM cells. Stimulation of iNKT cells by the α-galactosyl ceramide ligand can produce strong anti-tumour responses against MM cells in vivo. 15
NK cells
NK cells are a critical component of the innate immune system, and unlike T-cells, do not require antigen exposure to mount a response. Their effector function is determined by a balance between signals produced by inhibitory and activating receptors recognizing their respective ligands on tumour cells, or virally infected cells. Disruption of this balance enables immune evasion.
The most well characterized inhibitory receptors are the killer immunoglobulin-like receptor (KIR) superfamily, recognizing MHC (polypeptide related sequence) Class I antigens. Others include CD94/NKG2A (CD159a), ILT2, PD-1 and TIGIT. Activating receptors include NKG2D (CD314), which recognizes the MHC-related ligands MICA and MICB, the natural cytotoxicity receptors (NCRs), CD94/NKG2C (CD159c), CD16 (FcγRIIIA), the SLAM-related 2B4 (CD244) receptor, and DNAM-1 (CD226), which is regulated by TIGIT and CD96 (TACTILE). 16 Low-dose bortezomib was shown to increase expression of NKG2D and DNAM-1 ligands on myeloma cell lines, sensitising the cells to NK-mediated lysis, 17 suggesting a possible synergism between proteasome inhibition and NK cell therapies.
One of the most important inhibitory signals is HLA-E, which suppresses NK and T-cell activity through its interaction with NKG2A. Genetic variation in HLA genes means that around 40% express high levels of HLA-E and 60% express low levels. The impact of HLA-E expression was assessed in the MM Research Foundation CoMMpass study. Patients with high HLA-E expression were found to have a significantly shorter duration of progression-free survival (PFS) than those with medium or low-level expression. Mass cytometry in these patients also identified expansions of immune-suppressive T-cells with high levels of PD-1 and TIGIT expression, as well as a subset of Tregs, which were not seen in the groups with medium or low-level HLA-E expression. 18
MM cells and Tregs produce high levels of transforming growth factor (TGF)-β, which downregulates NK-activating receptors and impairs NK cytotoxicity. Elevated interleukin (IL)-10 levels inhibit IFN-γ and tumour necrosis factor (TNF)-α production, elevated IL-6, produced by both MM and bone marrow stromal cells (BMSCs) may inhibit NK activity, and elevated BM prostaglandin E2 levels inhibit activating signals via NCR, NKG2D and CD16. 16 Additionally, myeloid-derived suppressor cells (MDSCs) downregulate NK activity via the NKp30-activating receptor, membrane-bound TGF-β and TIGIT-mediated signalling.16,19,20
Presence of stress-induced MICA/B ligands on tumour cells activates NK cytotoxicity via NKG2D. Metalloproteinase-mediated cleavage of MIC generates soluble MIC ligands (sMICs). These cause internalization of NKG2D and other NK-activating receptors, leading to impaired cytotoxic activity. 21 MIC shedding has been seen in myeloma following exposure to doxorubicin and melphalan chemotherapy. 22 Surface plasma cell MICA expression is known to decrease with progression from MGUS to MM, 23 alongside other activating ligands. Conversely, there is evidence for upregulation of inhibitory ligands, for example, HLA Class I antigens. 24 In fact, MM cells from advanced disease states are so immunosuppressive to NK cells that they can evade killing by NK cells from normal healthy donors. 25
A further immune-evasive mechanism utilised by myeloma cells is surface expression of sialylated glycans, which bind to Siglecs (sialic acid-binding lectin receptor)-7 on NK cells (and Siglecs-9 on macrophages). Both treatment of MM cells with a sialytransferase inhibitor and use of NK cells lines with low Siglecs-7 expression, produces a significant increase in NK-medicated cell death. 26
Finally, NK cells in MM may show an exhausted phenotype, with downregulation of activating receptors, for example, NKG2D, NKp46 and DNAM-1 27 and increased expression of PD-1, leading to disrupted cytotoxicity and cytokine production, 28 and further increasing the ability of the malignant cells to escape immune surveillance.
Dendritic cells
DCs are professional APCs forming a critical link between the innate and adaptive immune system. High levels of circulating IL-6 in MM impairs the generation and function of DCs, stimulating CD34+ cells to differentiate into monocytic cells with potent phagocytic ability but no antigen-presentation activity. DCs isolated from MM patients have been shown to be unable to present tumour epitopes, unlike DCs from donors or those generated in vitro from CD14+ patient monocytes without exposure to excess IL-6. 29 Use of an IL-6 receptor alpha-chain knockdown DC vaccine in a murine model lead to increased production of tumour-specific CD8+ T-cells, increased cytokine production and improved PFS. 30
Hypersialylation of MM cells may additionally impair DC functioning, via binding to Siglec-7/-9 receptors on the DCs, leading to blunted T-cell activation, which can be ameliorated by inhibiting sialic acid expression. 31
Macrophages
Tumour-associated macrophages (TAMs) are derived by recruitment and activation of circulating monocytes by cytokines and chemokines produced by tumour cells and BMSCs. Activated macrophages are polarized into M1 or M2 macrophages. M1 macrophages are proinflammatory and produce high levels of TNF-α and IL-12, often in response to infections. TAMs usually resemble M2 macrophages, which are immunosuppressive, for example through expression of PD-L1, and stimulate angiogenesis favouring tumour growth. 32 Myeloma cells produce chemokines, for example, PGE2 which may attract and polarize macrophages to the M2 phenotype. IL-12, produced by M1 macrophages can downregulate myeloma cell angiogenesis and impair tumour growth. 33 Moreover, predominance of M2 macrophages has been linked to resistance to daratumumab/immunomodulatory drug (IMiD) combinations. 34 Strategies to repolarize macrophages could aid restoring responsiveness to daratumumab and other monoclonal antibodies.
MDSCs
This heterogenous group of cells is characterized by myeloid origin, immaturity and potent suppression of T-cell responses. MDSCs rapidly expand in response to infection, inflammation and malignancy, preventing massive T-cell over-activation and cytokine release. 35 This occurs partly by upregulation of inducible nitric oxide synthase (iNOS), leading to increased nitration of tyrosine residues in T-cells, preventing phosphorylation and inhibiting T-cell function. These cells are present at five-times normal levels in patients with newly diagnosed myeloma, providing yet another means of tumour escape from immune surveillance. Phosphodiesterase-5 (PDE-5) inhibitors, (e.g. sildenafil or tadalafil) downregulate iNOS production and can inhibit the suppressive effect of MDSCs on T-cell immunity. 36
BM stromal cells and adipocytes
BMSCs produce numerous cytokines and chemokines responsible for regulation of plasma cell migration, homing to the bone marrow niche, proliferation and survival, for example, CXCL-12 which binds plasma cell CXCR4 and facilitates migration, alongside IL-6, VCAM and CD44. 37 Elevated IL-6 is a poor prognostic feature, and high levels of the soluble IL-6 receptor correlate with poor responses to chemotherapy. 38 As mentioned previously, IL-6 has implications for T-cell, NK cell and DC immunity, and blocking IL-6 has been shown to improve tumour-specific responses in a murine model. 30
Therapeutics
Many of the agents used as standard of care in MM, alongside the novel targeted therapies have an immunomodulatory component to their mechanism of action. Here we will discuss the various classes of agents in turn, with a particular focus on the monoclonal and bispecific antibodies and cellular therapies under development. These are summarized in Figures 2 and 3, and key clinical trials in Table 1.
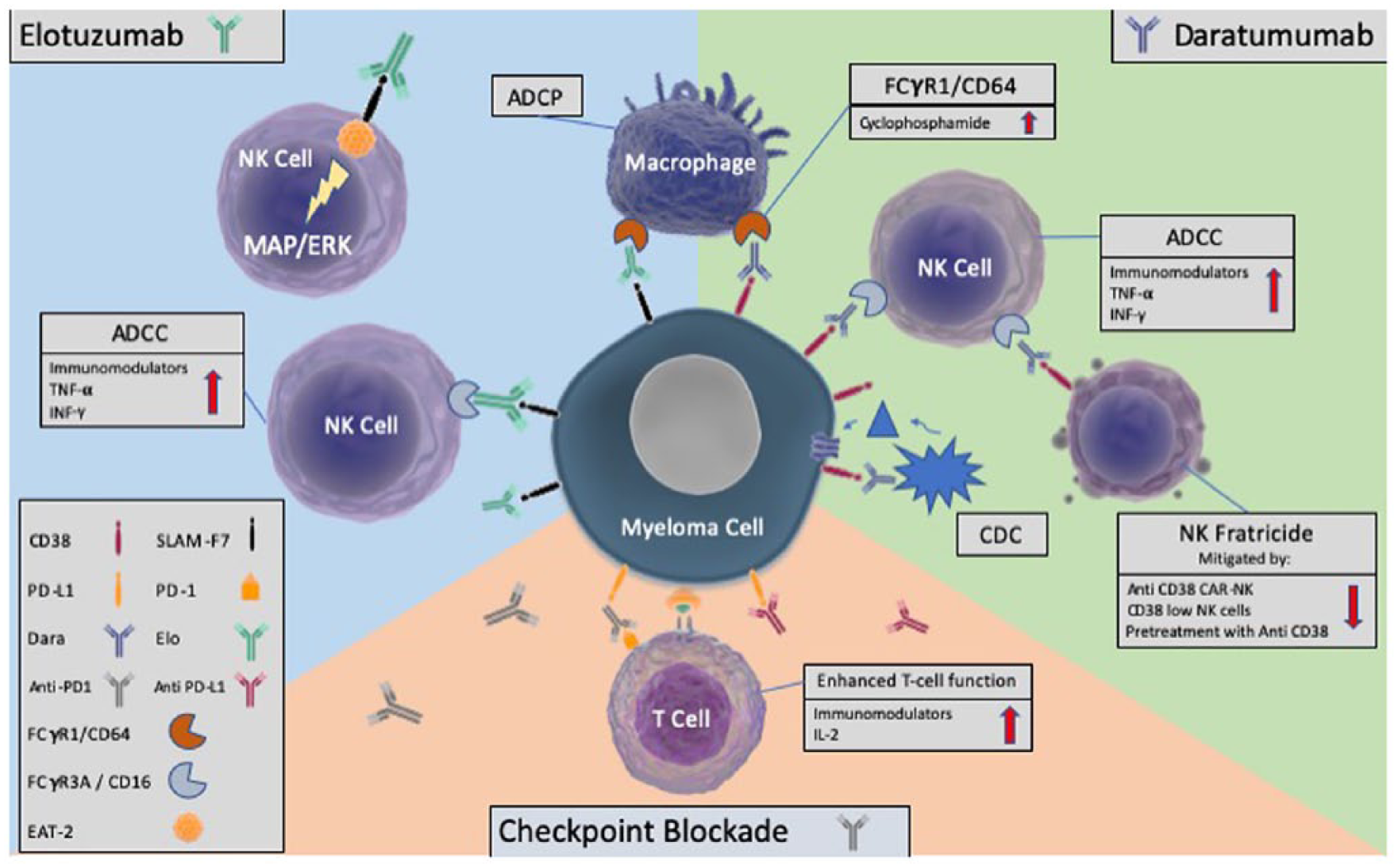
The activity of the monoclonal antibodies, daratumumab and elotuzumab, synergism with the immunomodulatory drugs and cyclophosphamide, and the role of checkpoint inhibition.

Therapeutic options for targeting myeloma epitopes, in this case, BCMA; CAR T-cells, bispecific T-cell engagers, bispecific antibodies and antibody–drug conjugates.
Clinical studies of myeloma immunotherapies.

AE, adverse event; ASCT, autologous stem cell transplant; BCMA, B-cell maturation antigen;
BiTE, bispecific T-cell engager; bort, bortezomib; CR, complete response; CRS, cytokine release syndrome; dara, daratumumab; dex, dexamethasone; elo, elotuzumab; GvHD, graft-versus-host disease; HR, hazard ratio; IMiD, immunomodulatory drug; IRR, infusion-related reaction; KIR, killer immunoglobulin-like receptor; len, lenalidomide; MIL, marrow infiltrating lymphocyte; MM, multiple myeloma; mon, months; MRD, minimal residual disease;
MTD, maximum tolerated dose; nCR, near complete response; NCT, ClinicalTrials.gov identifier; NDMM, newly diagnosed multiple myeloma; NK, natural killer; ORR, overall response rate; OS, overall survival; PD, progressive disease; PD-1, programmed cell death receptor 1;
pembro, pembrolizumab; PFS, progression free survival; PI, proteosome inhibitor; PR, partial response; RRMM, relapsed refractory MM; SAE, serious adverse event; sCR, stringent complete response; SD, stable disease; TCR, T-cell receptor; VGPR, very good partial response.
Immunomodulatory drugs
The IMiDs are thalidomide and its derivatives lenalidomide and pomalidomide. In vitro exposure of stem cells to IMiDs leads to growth and activation of DCs in a mouse model. 61 Lenalidomide and pomalidomide have been shown to enhance tumour antigen uptake and presentation by DCs, inhibit Tregs, and increase IL-2 and IFN-γ production, all leading to improved T-cell responses.62,63
IMiDs act via binding to cereblon, a component of the E3 ubiquitin ligase, resulting in ubiquitination and proteasome-mediated degradation of the Ikaros family zing finger protein transcription factors 1 and 3, and reduced transcription of MYC and IRF4, required for survival and proliferation. 64 Reduced levels of IKZF1/3 result in the upregulation of IL-2 and IFN-γ, stimulating NK growth and activity. A study using IL-2-primed peripheral blood mononuclear cells treated with thalidomide demonstrated significantly increased lysis of MM cell lines, independent of MHC class, and inhibited by depletion of CD56 positive cells, strongly suggesting NK-mediated killing. 65
Checkpoint inhibitors
PD-L1 is highly expressed on MM cells, as well as certain immune-suppressive cells within the tumour microenvironment (TME). Presence of PD-L1 impairs both T-cell and NK cell immunity, and levels of soluble PD-L1 have been shown to be independently associated with worse outcomes following upfront treatment. 66 A phase II study of 48 patients combined the PD-1 antagonist pembrolizumab with pomalidomide and dexamethasone in patients with RRMM (relapsed refractory MM), reporting 60% overall response rate (ORR), 19% very good partial response (VGPR) and 8% complete response (CR). 39 Studies of nivolumab, as a single agent however, failed to show efficacy. 67 Lenalidomide reduces the expression of both PD-L1 by myeloma cells and corresponding PD-1 by NK cells and cytotoxic T-cells, and has been shown to act synergistically with PD-1 blockade to improve NK and T-cell-mediated cytotoxicity in vitro, 68 offering a rationale for the enhanced efficacy seen compared with single-agent nivolumab. However, a phase III trial comparing pembrolizumab/lenalidomide/dexamethasone with lenalidomide/dexamethasone in front-line therapy was halted early due a treatment-related mortality of 3% in the intervention arm (KEYNOTE-185), 40 and a similar study comparing pembrolizumab/pomalidomide/dexamethasone with pomalidomide/dexamethasone (KEYNOTE-183) was found to have an unfavourable risk–benefit profile. 41 The United States Food and Drug Administration (US FDA) placed clinical trials using pembrolizumab on hold in July 2017 following the deaths in KEYNOTE-185, although this was partially lifted in December 2017 allowing ongoing recruitment in some studies. To date, PD-1 inhibition in MM has failed to replicate the successes seen in Hodgkin lymphoma and lung cancers.
Monoclonal antibodies
Monoclonal antibodies recognize specific tumour epitopes in order to generate an immune response and facilitate cell killing. Recent years have seen a marked expansion in monoclonal antibodies for use in MM.
Daratumumab is a fully humanized anti-CD38 monoclonal antibody that kills myeloma cells through antibody-dependent cellular cytotoxicity (ADCC), which relies upon binding of NK cell CD16 (FcγRIIIA), complement-dependent cytotoxicity (CDC) and antibody-dependent cellular phagocytosis (ADCP). Single-agent daratumumab yielded an ORR of 29% in RRMM. 42 The addition of lenalidomide has been shown to improve the ADCC of daratumumab in preclinical studies, and in our experience, low doses of cyclophosphamide augment macrophage-dependent ADCP, which will be discussed further under the heading of cyclophosphamide.69,70 Phase III trials of daratumumab with either lenalidomide/dexamethasone (POLLUX) or bortezomib/dexamethasone (CASTOR) in transplant-eligible patients have shown impressive responses in RRMM with a 12-month PFS of 83.2% versus 60.1% for standard of care, and 60.7% versus 26.9% respectively.43,44 Nontransplant candidates also have excellent responses with upfront daratumumab, in combination with bortezomib/melphalan/prednisolone in the ALCYONE trial, and lenalidomide/dexamethasone in the MAIA trial, presented as a late breaking abstract at ASH 2018.41,71 Based on the ALCYONE data, daratumumab is licensed upfront in elderly patients in Europe.
Elotuzumab is a humanized anti-SLAMF7 (signalling-lymphocyte-activating molecule F7) monoclonal antibody, also reliant upon NK-mediated ADCC and macrophage-mediated ADCP. Single-agent elotuzumab has not demonstrated significant efficacy; however, responses have been seen in combination with IMiDs or proteasome inhibitors, for example, the ELOQUENT-2 study with lenalidomide/dexamethasone reported a 27% reduction in risk of death or disease progression at 3 years in the elotuzumab arm. 45
Evidence of progressive NK cell impairment with disease progression suggests that these agents might be more efficacious upfront. Efforts to ameliorate the NK defects present in MM are described in a later section.
Bispecific T-cell engagers, bispecific antibodies and bispecific engagers
Bispecific antibodies offer several potential advantages over monospecific antibodies. They can simultaneously inhibit multiple survival and proliferation pathways or redirect immune cells to the tumour through expression of T-cell/NK-cell activating receptors, or increase tumour specificity via expression of more than one tumour-specific antigen, thereby reducing off-target toxicity.
Bispecific antibodies are classified as immunoglobulin (Ig)G-like, and non-IgG-like molecules, by the presence or absence of an Fc region. IgG-like bispecifics are capable of Fc-mediated functions, that is, ADCC, ADCP or CDC. They are larger, more stable with a longer half-life. Bispecific T-cell engagers (BiTEs) are a form of non-IgG-bispecific antibody, an scFv-based antibody, comprising only the variable heavy and light chain regions. They are smaller with improved tumour specificity and tissue penetration, but a shorter half-life necessitating frequent administration or continuous infusion. 72
BiTEs target a tumour epitope and a T-cell antigen, usually CD3. Recognition of the tumour epitope leads to T-cell engagement, activation, and cytotoxicity directed against the tumour cell, independent of TCR specificity, MHC complex expression, and antigen presentation. A phase I trial of the CD19-CD3 BiTE, blinatumomab in combination with salvage autologous stem cell transplant (ASCT) is due for completion in 2019 (ClinicalTrials.gov identifier: NCT03173430). MM cells typically do not express CD19, however various groups have isolated CD19-positive cells resembling B-lymphocytes, which give rise to MM cell colonies in vitro, and may act as myeloma stem cells,73,74 hence the rationale for blinatumomab use.
BCMA (B-cell maturation antigen) has been the focus of the majority of BiTEs to date.75–77 BCMA is a member of the TNF-receptor superfamily required for differentiation of B-cells into plasma cells. The BI 836909 BiTE, 76 showed promising in vitro data and was acquired by AMGEN as AMG-420. Early phase I results are encouraging: Of 35 patients, 6 CRs have been achieved, with minimal residual disease (MRD) negativity in all patients receiving a dose of 400 µg/d. Serious adverse events (SAEs) occurred in 49%, with infection the most common at 29%. There were three patients (9%) who developed cytokine release syndrome (CRS), which was grade 3 in one patient and grade 1 in the other two patients. 46 Another AMGEN BCMA BiTE under development is AMG-701, similar to AMG-420, but with an extended half-life. 78
Other bispecific antibodies under investigation include the tetravalent NK-engager AFM26, which targets MM BCMA and CD16a on NK cells to induce NK-mediated cell killing. 79 The requirement of CD16a for effective ADCC is hampered in approximately 80% of patients by the presence of a low affinity CD16a polymorphism. AFM26 interacts with NK cells with high-avidity, irrespective of this polymorphism. 80 An anti-CD16a/BCMA/CD200 antibody further increases specificity to tumour cells by binding both BCMA and CD200 on MM cells. 81
Antibody–drug conjugates
Antibody–drug conjugates are also in trial stages. GSK285916 is an anti-BCMA antibody linked to the antimitotic agent monomethyl auristatin F. In a phase I study of GSK285916 monotherapy in 35 patients with RRMM, ORR was 60% with CR or stringent complete response (sCR) in 14%, PFS was 12 months and median duration of response of 14.3 months. The most common adverse events (AEs) reported were thrombocytopenia (63% overall, 35% grade 3–4), blurred vision (51%, 3% grade 3, no grade 4 events), which was due to the effect of the monomethyl auristatin F, and cough (40%, grade 1–2 only). The most common SAEs were pneumonia or lung infection (15%) and infusion-related reactions (6%). Overall, 11% had AEs leading to trial discontinuation and 66% required dose-reductions.47,48,82 Both the US FDA and European Medicines Agency have since granted breakthrough status and PRIME designation respectively.
Cyclophosphamide
While cyclophosphamide is known to be an alkylator, it also has additional, valuable, immune-modulatory properties, which may enhance the activity of other immune based therapies. Metronomic dosing of cyclophosphamide selectively reduces Tregs, 83 allowing expansion of effector T-cells and enhanced NK activation, and may repolarize macrophages from the M2 to M1 phenotype.
The anti-CD38 monoclonal antibody daratumumab acts through ADCC and macrophage-dependent ADCP. In our experience, cyclo-phosphamide augments daratumumab-induced ADCP, possibly via upregulation of Fc receptor gamma 1 (CD64) and reduction in CD47 (a ‘don’t eat me’ antigen) expression by tumour cells. 69 This was confirmed by work in which macrophages were conditioned using culture media from myeloma cells exposed to low-dose cyclophosphamide, then incubated with MM cells and daratumumab. Enhanced daratumumab-induced ADCP of MM cells was demonstrated, alongside reduced MM CD47 expression. 70
Cellular therapies
The first cellular therapies took the form of allo SCT and DLIs. Chronic GvHD confers protection against relapse, 3 and DLIs can induce a graft-versus-tumour effect. 4 However, the increased rate of CR associated with allo SCT is offset by a high transplant-related mortality, partly attributable to GvHD. These data have prompted investigators to develop autologous T-cell therapies, aiming to preserve the anti-tumour effect but reduce or eliminate GvHD.
Adoptive T-cell therapies
Autologous T-cells are extracted from peripheral blood or BM (marrow infiltrating lymphocytes; MILs), then primed by antigenic stimuli, or genetically altered. The lymphocytes are expanded in vitro, usually by CD3/CD28 ligation, then reinfused into the patient.
Nonengineered
Studies of nonengineered T-cells have not demonstrated significant utility.84–86 MILs offer a theoretical advantage as their exposure to MM cells in the BM is increased, and tumour-specific populations are likely to be more prevalent. MILs also express the adhesion molecule CXCR4, to improve homing to the marrow. 49 A trial of MILs post-salvage ASCT in RRMM reported 27% CR, 27% partial response (PR) and 23% stable disease, although the proportion of response attributable to the MILs versus ASCT is uncertain. 49 Phase II trials are ongoing.
Engineered
Engineering a T-cell population enables delivery of far larger doses of tumour-specific T-cells. Autologous T-cells are transfected in vitro with viral vectors carrying the gene of interest, either a TCR, or a chimeric antigen receptor (CAR).
TCR T-cells
T-cells are engineered to encode TCRs against specific tumour antigen peptide-MHC complexes. One study using NY-ESO-1/LAG-1 TCR T cells in 20 patients reported responses in 80%, median PFS of 19.1 months and OS of 32.1 months. A total of 7 SAEs occurred (cytopenias, hypotension, hyponatraemia and GvHD) all of which resolved. Overall, three patients had diarrhoeal illnesses due to confirmed gastrointestinal GvHD. 50 TCR T-cells require the presence of certain HLA antigens and can be circumvented by tumour MHC-downregulation. They have been largely superseded by CAR T-cells.
CAR T-cells
CAR T-cells are engineered TCRs with an extracellular domain comprised of a single chain variable fragment of a tumour-specific monoclonal antibody. CAR T-cells can recognize a specific tumour epitope in the absence of MHC presentation and are independent of HLA type. In first generation CAR T-cells a transmembrane domain connects the antigen-recognition domain to the T-cell-activating CD3-zeta domain. Binding of the tumour epitope by the CAR therefore causes direct cytotoxic T-cell activation.
These first generation CAR T-cells were able to elicit a weak, short-lived T-cell response. Second and third generation CAR T-cells have an additional one or two, respectively, costimulatory domains (usually CD28 or 4-1BBB) which enhance cytokine production and cytolytic capacity. 87
The major toxicity of CAR T-cell therapy is CRS, due to the potent T-cell response generating a burst of IL-6, IFN-γ and other cytokines. This can cause hypotension, hypoxia, neurological dysfunction, multiorgan failure and fatality. Tocilizumab, an anti-IL6 receptor antagonist, and supportive care form the mainstay of management. 88
CD19 CAR T-cells are US FDA-approved for use in relapsed refractory B-cell precursor-ALL and non-Hodgkin lymphoma. Use in MM was trailed in order to target postulated CD19-positive myeloma stem cells. A total of 10 patients received salvage ASCT and CD19 CAR T-cells. ORR was 80% but all the patients progressed with a median PFS of 185 days. 51 A study using 10 times the dose of CD19 CAR T-cells post front-line ASCT is ongoing (ClinicalTrials.gov identifier: NCT02794246), and another using CD19 and BCMA-CAR T-cells post-ASCT has reported an ORR of 100% in nine evaluable patients, with increased MRD negativity post-CAR T-cell infusion (37.5% post-ASCT to 66.7% post-CAR T-cells). 89
Other antigen targets have been trialled in myeloma with varying degrees of success. CD44v6, CD70, CD38, CD138, kappa light chain, SLAMF7, and GPRC5D (G protein-coupled receptor class C group 5 member D) 90 are examples. The challenge of target selection is identifying an epitope which is present in high concentration on MM cells in a significant proportion of myeloma patients, but not present on normal haematopoietic cells or other tissues. Very few epitopes meet these criteria. CD38 for examples, is present on malignant and benign plasma cells, T-cells, B-cells, NK cells, DCs, erythrocytes and many others. Targeting such antigens leads to ‘on target/off tumour’ effects. Drent and colleagues optimized the affinity of their CD38-CAR T-cells to bind to strongly CD38-positive myeloma cells, but to have inadequate affinity to bind to cells with lower levels of expression. This produces T-cell-mediated MM cell lysis with the sparing of normal haematopoietic cells. 91
The most promising work has been using anti-BCMA-CAR T-cells. BCMA is expressed by late memory B-cells, plasmablasts, mature plasma cells and myeloma cells, but not haematopoietic stem cells or nonhaematopoietic cells. 92 The first-in-human clinical trial of anti-BCMA-CAR T-cells, run by the National Cancer Institute (NCI), used fludarabine/cyclophosphamide followed by a CAR T-cell infusion in 12 patients with RRMM. Of these, 10 received a low dose, obtaining mostly stable disease. Of the two patients receiving the higher dose, one achieved stringent CR for 17 weeks and the other, VGPR for 66 weeks, but both had severe CRS. 53
Bluebird Bio received breakthrough status by the US FDA for the bb2121 BCMA-CAR. 21 patients with RRMM, two thirds with adverse cytogenetics, were infused with bb2121, an anti-BCMA-CAR expressing the same single chain Fc portion as the NCI trial used, but with a 4-1BB costimulatory domain. The latest results were presented at ASCO 2018. At doses of ⩾150 × 106 the ORR was 94% with a CR of 56%. Overall, 71% of patients had CRS which was generally mild, and only one fifth required tocilizumab. Median PFS has not yet been reached. Rates at 6 and 9 months are 81% and 71% respectively, and a deepening of response over time has been noted, with several patients converting to CR from initial VGPR. 54 A modification of the bb2121 CAR, termed bb21217, is treated with the phosphoinositide-3 kinase inhibitor bb007 during ex vivo culture. This enriches the product for memory-like T-cells, leading to increased persistence and efficacy of the CAR T-cell product. Early results have shown responses in six out of seven evaluable patients, with MRD negativity demonstrated in all of the three responses tested. 55
Legend Biotech’s LCAR-B38M anti-BCMA-CAR has two different heavy chain variable domains which recognize separate epitopes of the BCMA antigen. Initial data showed an ORR of 100% at a median 17 months follow up in 19 evaluable patients. 56 The LEGEND-2 trial (ClinicalTrials.gov identifier: NCT03090659) recently reported results on 57 patients who received LCAR-B38M CAR T-cells. Grade 3 AEs occurred in 65%, with leukopenia (30%) and thrombocytopenia (23%) most commonly reported. CRS occurred in 90% but the majority of cases were grade 1–2 (83%). ORR was 88% with a CR of 68% and 63% of patients achieving MRD negativity. At a median follow up of 8 months, median PFS was 15 months. 57
Finally, preclinical studies are trying to overcome the challenge of heterogenous BCMA expression and antigen escape which limit effectiveness of current BCMA targeted therapies. APRIL (A proliferation-inducing ligand), a member of the TNF superfamily of ligands, binds simultaneously to both BCMA and TACI (transmembrane activator and calcium modulator and cyclophilin interactor), both of which are consistently upregulated on myeloma cells. Schmidts and colleagues have generated monomeric and trimeric APRIL CAR T-cells, of which the trimeric form (TriAPRIL-CAR) displayed superior binding to soluble BCMA and TACI and lead to tumour eradication in a murine MM model. 93
NK cells
NK activity is tightly regulated by the overall balance between numerous activating and inhibitory stimuli. KIR-ligand mismatch, indicating nonself, is associated with reduced relapse rate following T-cell depleted alloSCT. 94 An IgG monoclonal antibody directed against three different KIRs (IPH2101) augments NK-mediated lysis of HLA-C-expressing tumour cells in vitro, 95 but failed to show clinical benefit in a phase II trial in smouldering myeloma. 96 A recombinant version, lirilumab is presently being tested in combination with elotuzumab (NCT02279394).
Daratumumab relies partly upon NK cells for ADCC, and efficacy is improved by the addition of IMiDs, which upregulate NK activities. 43 However, due to CD38 expression on NK cells, daratumumab significantly depletes NK cells during treatment and for up to 6 months after. 97 Several groups are working on NK cell adoptive transfer to enhance the activity of daratumumab, using approaches to mitigate fratricide such as CD38-deletion, use of low CD38 expressing NK cells or pretreatment of NK cells with Fab fragments to block CD38.98,99
The anti-SLAMF7 monoclonal antibody elotuzumab induces ADCC and also activates NK cells via an indirect mechanism dependent on the SLAMF7-associated adaptor protein, EAT-2. Elotuzumab binding to the NK cell SLAMF7 engages EAT-2, activating the MAPK/Erk pathway. As myeloma cells lack EAT-2, elotuzumab-binding does not stimulate the proliferation of MM cells. Again, elotuzumab showed improved clinical benefit when administered with lenalidomide, 45 possibly due to further upregulation of NK activity.
Use of NK cells to stimulate a graft versus MM effect via KIR-mismatch is also being tested. A phase I study in RRMM used multiple NK cell infusions with lenalidomide or bortezomib-based regimens without significant toxicity. Disease stabilized in four out of five patients, with one response maintained after 12 months. 58 Another group used cord-derived NK cells with lenalidomide and ASCT in 33 patients, producing 83% CR or VGPR at 3 months, with no toxicities reported. 59
CAR NK cells offer an additional possible avenue. The combination of daratumumab with CD38-negative NK cells engineered with the CS1 CAR (SLAMF7) is under investigation, in order to target two myeloma-specific epitopes while avoiding daratumumab-induced NK destruction. 100 A CD38 CAR NK cell line is currently being explored, using an affinity optimized scFv, similar to that reported by Drent and colleagues 91 to facilitate differentiation between CD38-positive NK cells and myeloma cells respectively. In vitro results, presented at ASH 2018, have shown high cytotoxicity against MM cells, with in vivo results awaited. 101 Initial murine model data regarding an NKG2D-CAR NK cell line have also recently been presented, with further work ongoing. 102
Vaccines
Vaccines are designed to elicit production of tumour-directed T-lymphocytes by the host immune system. The greatest success has been seen after allo- or ASCT, when disease burden is low, and lymphoid reconstitution is occurring with low Treg levels and expansion of CD8+ cells. 103 Vaccines can be based on specific, or multiple tumour peptides, whole cells, or incorporating DCs as potent APCs. DCs loaded with/fused to tumour antigens may bypass the DC functional defects seen in myeloma and have provided the most promising results to date. For example, a DC–tumour cell fusion vaccine in RRMM produced a significant rise in myeloma-specific T-cells and disease stabilization for at least 2 months in 70%, with serial vaccination shown to deepen responses post-ASCT. 60 Hypersialyation is a feature of MM 104 which impacts DC function. Sialyltransferase inhibition to enhance DC response may provide a possible means to improve responses to vaccination; 31 however, given the profound impact of myeloma on the immune system, whether this approach will prove clinically useful remains uncertain.
Conclusion
Our understanding of the mechanisms that underpin immune evasion by MM cells has increased significantly in recent years, and the development of novel agents that disrupt and target these mechanisms represents a paradigm shift in how we approach the treatment of myeloma.
A wealth of new treatments exists, although in most cases their use is currently limited to the relapsed refractory setting, with many still in clinical trials. Given the worsening immune dysfunction that occurs with disease progression, it is likely that the greatest benefit will be seen with earlier treatment. This may be particularly relevant for patients with high-risk genetic features, those with a suboptimal response to front-line therapy, early relapse within 1 year or those who remain MRD-positive despite intensive therapies, for whom improved treatment options are desperately needed. The optimal sequence of available therapies is also unknown, and how best to manage patients who are refractory to or relapse after front-line therapy involving immunomodulatory agents has recently been highlighted as an area of unmet clinical need. 105
The most promising results to date have been obtained with CAR T-cell therapies. However, these treatments are associated with significant challenges including treatment delays, potential for severe toxicity, manufacturing issues, logistics required to administer such therapies, and the significant associated costs. These challenges may ultimately limit widespread application of CAR T-cell therapies, therefore interest in the development of ‘off-the-shelf’ alternatives with increased tolerability is growing.
Ultimately, we hope that more effective primary therapy may produce long-lasting, durable remissions, or possibly even cure in a significant fraction of patients.
Footnotes
Author contributions
DS performed the literature review and wrote the manuscript. KL designed Figure 1. MG designed Figures 2 and 3. MOD conceived the review and critically reviewed the manuscript.
Conflict of interest statement
MOD is a Founder and Director of Onkimmune and has received research support from Janssen, Celgene and Bristol Myers Squibb.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.