
Abstract
Kimura’s disease (KD) is a rare, benign disorder characterized by subcutaneous masses with regional lymph-node enlargement. It is considered to be due to chronic inflammation of unclear etiology. Most cases have been reported in young, 20–30-year-old men of Asian descent. The diagnosis of KD is based on pathological features and elevated immunoglobulin E levels. Characteristic pathological features include intact lymph-node architecture, florid germinal center hyperplasia, extensive eosinophilic infiltrates, and proliferation of postcapillary venules. However, these features can also be seen in Hodgkin’s disease or T-cell lymphoma, therefore, cases presenting as KD pose a diagnostic challenge. We report a case series of two cases with suspected KD at initial presentation, with one patient eventually diagnosed with Hodgkin’s disease after clinical progression. The first case was a 45-year-old Asian man who presented with bilateral thigh masses and significantly enlarged inguinal lymph nodes. The histopathology was characteristic and the patient had stable disease on treatment with cetirizine for 20 months. The second case was a 29-year-old African-American man who had progressive enlargement of the right neck lymph nodes extending into the mediastinum, with the original biopsy suggestive of KD. An initial search for Reed–Sternberg cells using immunohistochemical staining for CD15 and CD30 was negative. However, the patient developed neurological symptoms corresponding to tumor extension to the cervical and thoracic neural foramina. A repeat biopsy showed a lack of nodal structure and atypical large cells that were positive for CD30 staining. The patient was treated with chemotherapy with good response. We emphasize the importance of following the clinical course to render an accurate diagnosis. Both cases showed extensive eosinophilic infiltration and other KD-like pathological features. However, KD is rare; not missing a malignant diagnosis lies in high clinical suspicion and repeated exhaustive work up.
Introduction
Kimura’s disease (KD) is a rare, benign disorder associated with chronic inflammation of unclear etiology. 1 It presents as slowly enlarging, painless subcutaneous masses, predominantly in the head and neck region, and is often accompanied by regional lymphadenopathy.2,3 Insidious in onset, it follows an indolent clinical course. Most of the reported cases are in young men aged 20–30 years, mostly from the Far East, although sporadic cases have also been reported in all ethnic groups. 1
The diagnosis of KD is mainly based on characteristic pathological features, which include florid germinal center hyperplasia, extensive eosinophilic infiltrates, and proliferation of the postcapillary venules. 4 Accurate diagnosis is aided by the presence of markedly elevated peripheral eosinophil count and serum immunoglobulin E (IgE) levels.2,3 However, the above pathological features are not pathognomonic for KD, and a number of malignant conditions, such as Hodgkin’s disease and T-cell lymphoma, can closely mimic these findings. 1
Herein, we present two cases to illustrate this diagnostic challenge. One case presented with an initial diagnosis of KD, which eventually turned out to be a ‘KD mimic’, with a true diagnosis of Hodgkin’s disease. These cases underscore the importance of high clinical suspicion for underlying malignant diagnosis if a patient shows clinical progression.
Case 1
A 45-year-old Chinese-American man presented with a painless, progressively enlarging mass in the left upper thigh over 3–4 months. He denied fever, night sweats, or weight loss. His past medical history was significant for glomerulonephritis at the age of 14 years with full recovery following treatment and left submandibular lymph-node resection at the age of 35 years. On physical examination, a large palpable mass was found in the left medial upper thigh proximal to the inguinal area, it was nontender and nonerythematous. A computed tomology (CT) scan showed bilateral anterior thigh masses with the left larger than the right (Figure 1). It was confirmed on magnetic resonance imaging (MRI) as a multinodular, slightly serpiginous, 10 cm × 5 cm × 2.5 cm mass, involving the subcutaneous fat, without deep structure involvement, with enhancement after intravenous gadolinium. The left inguinal and right submandibular lymph nodes were also enlarged. The laboratory tests showed a white blood count (WBC) of 8000/mm3 with 20% eosinophils and an absolute eosinophil count of 1640/mm3. His IgE levels were also elevated at 8.77 × 105 IU/L (normal range < 11.1 × 103 IU/L). The hemoglobin and platelet counts as well as the comprehensive metabolic panel (CMP) were within normal range.
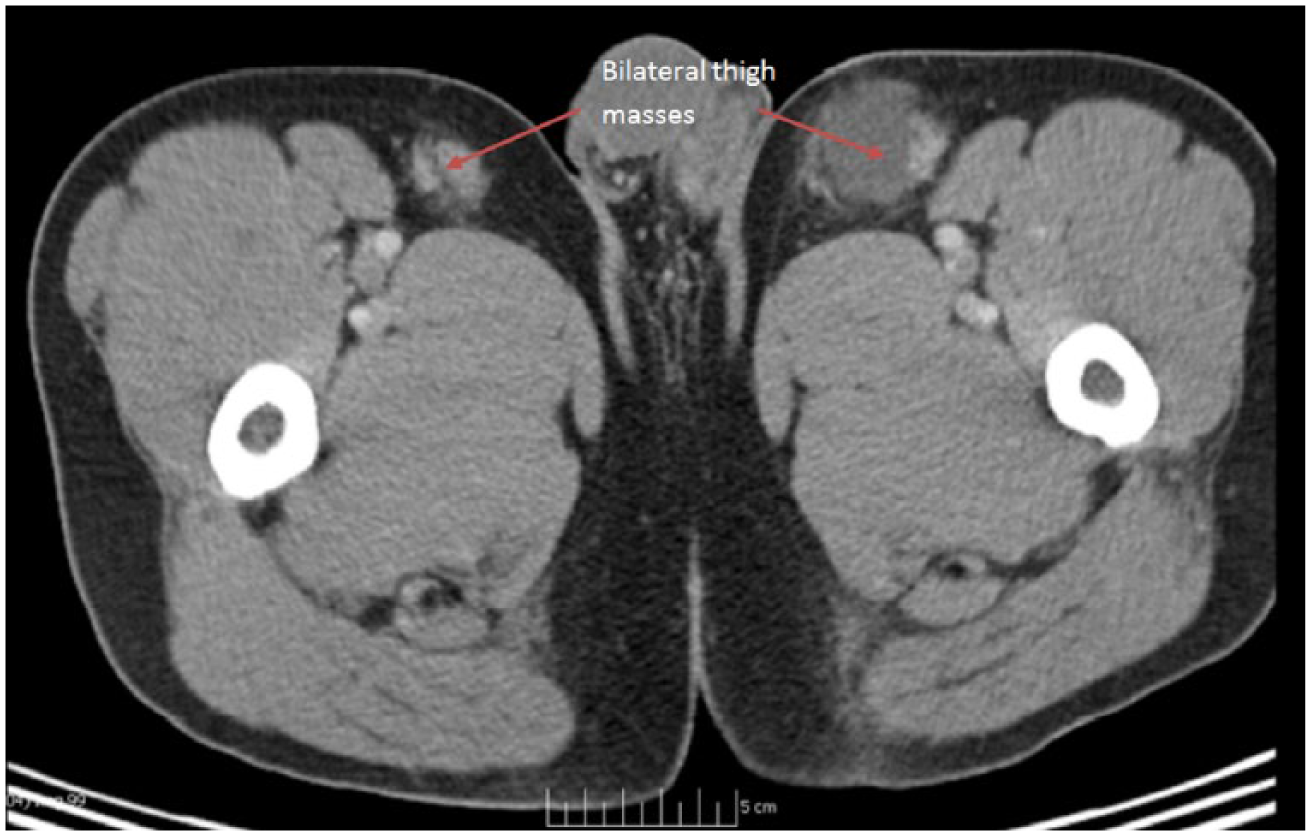
Computed tomology scan showing bilateral anterior thigh masses, the left bigger than the right. (Arrows demonstrate bilateral thigh masses.)
An incisional biopsy of the left thigh mass showed preserved lymph-node architecture with follicular and focal paracortical hyperplasia. The interfollicular region contained scattered eosinophils and many endothelial venules. The perinodal fibroadipose tissue was extensively infiltrated by a polymorphous inflammatory infiltrate, including reactive lymphoid follicles, and numerous eosinophils which focally formed microabscesses (Figure 2). Besides, proliferation of post capillary venules was also seen. There was predominant deposition of IgE within the germinal centers. No Reed–Sternberg-like cells were identified. Flow cytometry showed polytypic B cells and T cells with no antigenic loss or aberrant expression. This confirmed the diagnosis of KD.
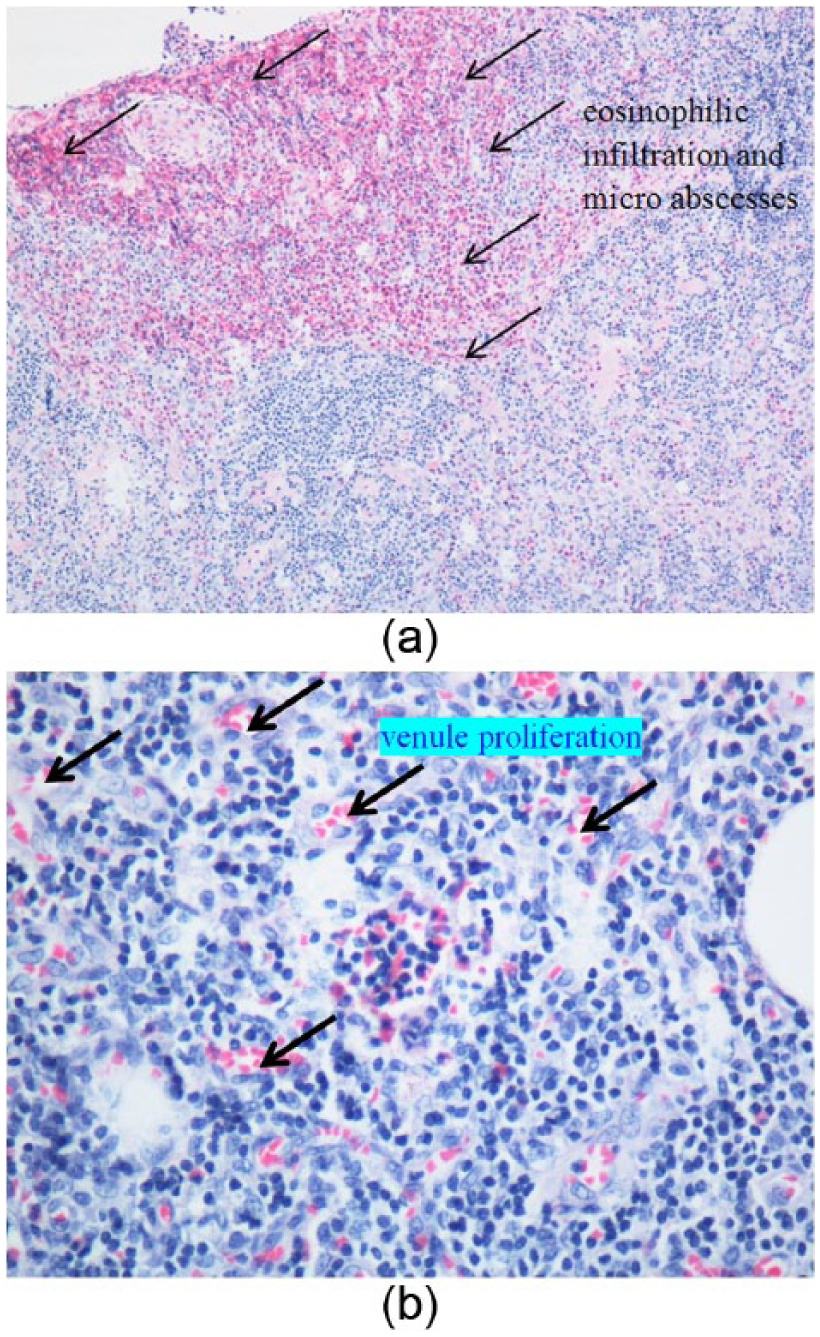
(a) Lymph-node biopsy from case 1 showing extensive eosinophilic infiltration in the subcortical region forming eosinophilic microabscesses (hemotoxylin and eosin × 100). (Arrows demonstrate eosinophilic infiltration and microabscesses.) (b) Lymph node biopsy showing extensive postcapillary venule proliferation (hemotoxylin and eosin × 400). (Arrows demonstrate venule proliferation.)
The patient was started on cetirizine 10 mg once daily. He was followed up every 3 months for the first 6 months, and then once every 6 months. He remained asymptomatic, and at the 20 months follow up, the left thigh palpable mass appeared to be stable whereas the right thigh mass became non-palpable.
Case 2
A 29-year-old African-American man presented with progressive swelling of the right side of his neck for more than 3 months. It was associated with mild pain but without any fever, night sweats, or weight loss. An MRI scan of the neck showed massively enlarged right neck lymph nodes extending to the supraclavicular area, upper mediastinum, and also through the C6, C7, T1, and T2 vertebral neural foramina. The mass showed enhancement with gadolinium.
A chest X-ray showed right hilar and right paratracheal lymphadenopathy. The patient’s complete blood counts showed hemoglobin of 11.7g/dl, WBC of 8600/mm3 with 6% eosinophils (absolute eosinophils of 516/mm3), and normal platelet count. His CMP was normal. A CT-guided core needle biopsy showed reactive lymphoid infiltrate with small vessel proliferation associated with numerous eosinophils. No cells with Reed–Sternberg phenotypes were identified, and staining with CD15 and CD30 was negative. The differential diagnosis was KD and angiolymphoid hyperplasia with eosinophilia (ALHE). Approximately four months later, the patient developed new symptoms including pain in the right axilla with numbness and tingling in his right arm. Repeat imaging revealed stable lymphadenopathy. A repeat incisional biopsy showed eosinophil-rich lymphohistiocytic infiltrate associated with dense fibrosis. No nodal architecture was identified. Rare CD30 (+) large atypical cells with Reed–Sternberg-like features were identified. Flow cytometry showed polytypic B- and T-cells with no antigenic loss or aberrant expression. CD4: CD8 ratio was normal 1:1. Immunohistochemical studies revealed a classical Reed–Sternberg profile (CD30 positive, CD15 variable, and weak expression of PAX-5, OCT-2, and BOB-1). CD1A-positive Langerhans cells were moderately increased within the inflammatory stroma. There was no evidence of clonal IgH rearrangement or T-cell receptor rearrangement. A diagnosis of classical Hodgkin’s lymphoma, nodular sclerosis subtype, was suggested. A positron emission tomography (PET) CT performed 10 weeks after the initial presentation showed a marked increase in the size of the supraclavicular lymph nodes, and increased fluorodeoxyglucose (FDG) uptake in all the above lymph nodes as well as in retroperitoneal lymph nodes, which were subcentimeter in size.
The patient was treated with glucocorticosteroids initially with marked improvement in his symptoms of right upper extremity pain, followed by chemotherapy with Adriamycin® (doxorubicin), bleomycin, vinblastine, dacarbazine regimen.
Discussion
The pathophysiology of KD remains uncertain. 5 Increased production of IgE and cytokines such as tumor necrosis factor-alpha, interleukin (IL)-4, IL-5, and IL-13 at the lesion site has been observed. 6 OKT4-positive granuloma T-cells have been accounted for the production of natural mediators for tissue eosinophilia in KD. This cytokine dysregulation is the result of the predominance of T helper cell 2 and Tc1 cells. 7 However, no specific antigen trigger has been established yet. In a study, overexpression of IL-21 and phosphorylated extracellular signal-regulated kinase (pERK1/2) was observed in 18 patients with KD compared with age- and gender-matched controls. The pERK1/2 was also suggested to bear prognostic significance as patients with pERK 1/2 had higher disease-free survival on multivariate analysis. 8
The pathological features of KD have been well described in a comprehensive review. 4 Some of the most common findings are listed as ‘constant features’, including intact nodal architecture, extensive reactive lymphoid infiltrates, postcapillary venule proliferation, and infiltration with numerous eosinophils accompanied by mast cells, histiocytes, and Langerhans cells. Some other less common features are described as ‘frequent features’, which are sclerosis, polykaryocytes, vascularization of the germinal centers, proteinaceous deposits, necrosis of the germinal centers, eosinophilic abscess, and a reticular IgE deposition within germinal centers. Both of our cases share a number of features that are ‘constant features’, while the first case also displayed some of the ‘frequent features’. However, none of the above features are pathognomonic for KD: they can be found in numerous other benign and malignant disorders, including ALHE, Hodgkin’s lymphoma, angioimmunoblastic T-cell lymphoma, Langerhans cell histiocytosis, florid follicular hyperplasia, Castleman’s disease, dermatopathic lymphadenopathy, allergic granulomatosis of Churg–Strauss syndrome, lymphadenopathy of drug reactions, and parasitic lymphadenitis. 1 With such pathological descriptions, it is challenging to rule out more common malignant diagnoses, namely Hodgkin’s disease and T-cell lymphoma.
Some characteristic imaging findings favoring KD are the appearance of ill-defined subcutaneous masses, which are homogeneously isodense to hypodense without necrosis, calcification, or cystic degeneration on CT. 9 On MRI, these masses appear hypointense to isointense on T1-weighted images and hyperintense on T2 images.9,10 The level of enhancement and intensity varies on both CT and MRI images due to the varying degrees of fibrosis and vascular proliferation. On 18F FDG PET, KD lesions show remarkable uptake in the regions mimicking other inflammatory or neoplastic disorders, such as tuberculosis, lymphoma, or metastatic lymphadenopathy. 11
Patients with KD demonstrating unusual manifestations such as generalized lymphadenopathy or a pulmonary hilar tumor on 18F FDG PET have also been described in the literature.12,13 Cases of KD mimicking Hodgkin’s disease or vice versa have been reported due to the close resemblance on histology and imaging.11,14 Although in most cases of KD, the tumor masses are predominantly located in the head–neck area, 1 our case of KD showed the involvement of bilateral thighs. Since Hodgkin’s disease also affects head and neck region frequently, location alone does not favor KD over HD. Besides the head and neck, cases with axillary lymph node and breast involvement have also been reported.15,16 More extensive patterns of disease should prompt consideration of an alternative diagnosis. The key is to follow the clinical course with a low threshold for extensive work up to rule out malignancy in patients who show clinical progression. In the second case, the presence of atypical large cells as well as the lack of lymph-node architecture raised the suspicion and repeated work up eventually established a diagnosis of Hodgkin’s disease.
Our first case also exemplifies other features typical of KD such as peripheral eosinophilia and elevated IgE.2,3 Perceivably, his left mandibular lymph node which was resected while he was young, might have been the first manifestation of KD, and his adolescent glomerulonephritis could be an autoimmune manifestation associated with KD.2,3
Treatment of KD is mainly for cosmetic reasons or for the preservation of functioning while preventing recurrences. 17 Variable success has been obtained with surgical excision, drug therapy with antihistamines and glucocorticosteroids, low-dose radiation, cytotoxic agents, and targeted therapy.18–20 Higher levels of c-kit and platelet-derived growth factor receptor-α, similar to hypereosinophilic syndrome, were reported in patients with KD which suggests the possibility of imatinib in these patients. 21 However, this has not been confirmed clinically.
Conclusion
KD is a benign disorder characterized by subcutaneous soft tissue masses or lymph-node enlargement with an indolent clinical course. It can mimic malignancy and thus these patients warrant close follow up and a low threshold for the repeat work up.
Footnotes
Acknowledgements
The authors would like to thank Michael Simon for his assistance in proofreading and formatting this paper.
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
The authors declare no conflicts of interest in preparing this article.