
Abstract
B-cell lymphoma 2 (BCL2)-type proteins are key regulators of the intrinsic or mitochondrial pathway for apoptosis. Since escape from apoptosis is one the main ‘hallmarks of cancer’, BCL2 inhibitors have emerged as promising therapeutic agents for diverse lymphoid malignancies, particularly chronic lymphocytic leukemia (CLL). Multiple clinical trials have shown efficacy of these agents in patients with relapsed/refractory disease with a favorable toxicity profile. Moreover, some clinical trials indicate that combination with monoclonal antibodies and other novel agents may enhance their effect.
Introduction
Treatment for patients with chronic lymphocytic leukemia (CLL) has changed remarkably in the last few years [Rai and Jain, 2016]. For decades, it was based on alkylating agents and similar drugs with modest efficacy and no impact on survival. The introduction of fludarabine and other purine analogues led to a significant improvement in complete remission rate, response rate and progression free survival [Rai et al. 2000], which was further substantiated by the addition of monoclonal antibodies, particularly those targeting CD20 [Hallek et al. 2010]. Currently, the outlook of patients with CLL is becoming brighter with the advent of modern targeted therapies, including second-generation monoclonal antibodies [Wierda et al. 2010; Goede et al. 2014], immune modulators [James et al. 2014], Bruton’s tyrosine kinase (BTK) inhibitors [Woyach et al. 2014a; Brown et al. 2014], phosphoinositide 3’-kinase (PI3K) inhibitors [Brown et al. 2014] and B-cell lymphoma 2 (BCL2) antagonists [Ng and Davids, 2014].
In this manuscript, we will review the scientific basis for the use of BCL2 inhibitors as a treatment for lymphoid malignancies with a special focus on CLL.
The role of apoptosis in programmed cell death
Apoptosis is a very important form of programmed cell death that has been conserved throughout evolution. This cellular process is executed by a number of proteases, called caspases, which can be activated in two ways: the extrinsic and the intrinsic pathways [Brahmbhatt et al. 2015; Juin et al. 2013]. The extrinsic pathway is triggered by cell-surface death receptors (such as Fas) coupled to extracellular signals, and the intrinsic or mitochondrial pathway is regulated by the BCL2 family of proteins.
The effectors of this intrinsic pathway are two multi-domain pro-apoptotic molecules called BAX and BAK that, when activated, acquire the ability to insert into mitochondrial membranes, create pores, and trigger mitochondrial outer membrane permeabilization (MOMP). MOMP releases cytochrome c from inside the mitochondria into the cytoplasm which, in turn, activates the caspases and subsequent cell death [Davids and Letai, 2012]. In addition, BAX and BAK are tightly regulated by a series of pro-apoptotic and anti-apoptotic proteins. When in stress or damage, cells generate pro-apoptotic proteins with a single active domain called BH3. These single-domain proteins could be classified as activators, such as BID or BIM, that are able to activate BAX/BAK directly; and sensitizers, such as BAD, NOXA or PUMA, that inhibit multi-domain anti-apoptotic molecules [e.g. BCL2, BCL-XL, myeloid cell leukemia 1 (MCL1), BCL-W, BFL-1]. Among these anti-apoptotic proteins, we will focus our attention on three of them, namely BCL2, BCL-XL and MCL1. These multi-domain proteins block apoptosis through two different mechanisms, either by directly inhibiting BAX/BAK, but also by sequestering pro-apoptotic proteins (Figure 1) [Davids and Letai, 2012].
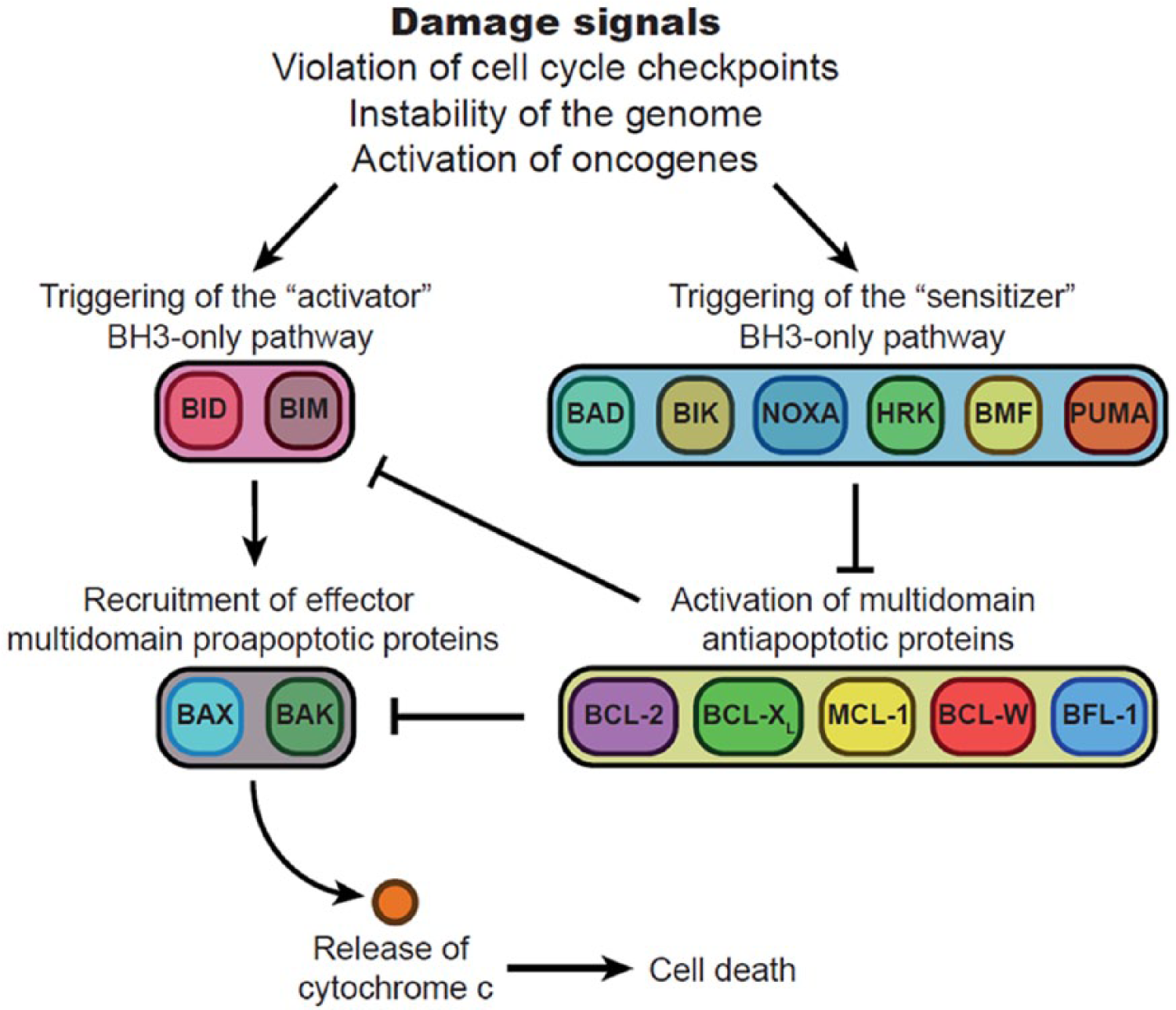
Role of BCL2 and related proteins in the regulation of apoptosis. Signals of damage or derangement (violation of checkpoints, instability of the genome, activation of oncogenes) induce overexpression of pro-death BCL2 homology 3 (BH3)-only proteins. These proteins are functionally subdivided into activator (left) and sensitizer (right) protein families. Activator BH3-only proteins such as BID (BH3-interacting domain death agonist) and BIM (BCL2-like 11) induce effector multi-domain proapoptotic protein overexpression such as BAX (BCL2-associated X) and BAK (BCL2 antagonist/killer 1). This, in turn, leads to mitochondrial changes and apoptosis. Multi-domain anti-apoptotic proteins such as BCL2 oppose this process by sequestering activators, which limits contact with effectors, or by sequestering activated effectors. Sensitizer BH3-only proteins such as BAD (BCL2-associated agonist of cell death) act as selective inhibitors of anti-apoptotic proteins. Arrows and blunted arrows represent activation and inhibition, respectively.
In summary, at any given time the fate of a particular cell results from a delicate balance between pro-apoptotic and anti-apoptotic proteins. If there is an excess of pro-apoptotic signals, BAX and BAK are activated and the cell dies. If otherwise, the cell detects pro-survival stimuli, anti-apoptotic proteins like MCL1 or BCL2 prevail and the cell survives.
The role of B-cell lymphoma 2 in normal and malignant lymphoid cells
When a normal lymphocyte is inactive, pro-apoptotic and anti-apoptotic proteins are balanced, thus promoting cell survival. In contrast, if the cell detects any kind of danger, such as radiation, chemotherapy, oxidative stress or cytokine deprivation, a number of BH3-only pro-apoptotic molecules are generated which, on the one hand block BCL2 and other anti-apoptotic molecules, and on the other hand activate BAX/BAK directly. The net effect of this BAX/BAK activation is MOMP, which leads to cytochrome c release into the cytoplasm, caspase activation, and cell death [Anderson et al. 2014].
Since apoptosis is crucial for cell homeostasis, malignant cells try as much as they can, and CLL cells are no exception, to evade this cellular process. Indeed, escape from apoptosis has long been recognized as one of the main ‘hallmarks of cancer’, contributing to both oncogenesis and drug resistance [Hanahan and Weinberg, 2000, 2011]. In order to accomplish this goal, CLL cells can do several things (Figure 2):
Upregulate anti-apoptotic proteins such as BCL2 or MCL1. BCL2 overexpression is mainly caused by hypomethylation of the BCL2 gene promoter [Hanada et al. 1993] and deletion/downregulation of miR-15/16 genes [Cimmino et al. 2005], whereas MCL1 overexpression is induced and maintained by marrow stromal cells [Pedersen et al. 2002].
Disrupt tumor suppressor genes, such as TP53, thereby decreasing the activation mediated by NOXA or PUMA (p53-upregulated modulator of apoptosis).
Downregulate pro-apoptotic proteins such as BAX or BAK or, more precisely, increase the BCL2/BAX ratio [Pepper et al. 2008].

Control of apoptosis by the BCL2 family of proteins. In normal mature B-cells (upper panel), BCL2 maintains cellular viability by blocking apoptosis. When a biological stress signal appears (middle panel), such as DNA or microtubular damage, cytokine deprivation or oxidative stress, the BH3 (BCL2 homology 3)-only proteins are activated. These proteins allow apoptosis to occur by binding to and inactivating BCL2 and related anti-apoptotic proteins. Some BH3-only proteins can directly activate BAX (BCL2-associated X) or BAK (BCL2 antagonist/killer 1) in malignant lymphoid cells (lower panel), stress-induced apoptosis may be impaired through a number of different mechanisms including overactivity of the anti-apoptotic BCL2 proteins (lower panel, upper row), reduced BH3-only protein expression via TP53 gene disruption (tumor protein 53, lower panel, middle row) and loss of BAX or BAK (lower panel, lower row). Black arrows and black blunted arrows represent activation or inhibition, respectively. Grey arrows and grey blunted arrows represent weakened activation or inhibition, respectively, due to biological influences acting upon them.
Numerous studies in CLL have evaluated all these mechanisms in vitro and in vivo. Indeed, patients with higher levels of BCL2 have a shorter survival compared with those with lower BCL2 expression. Equally, patients with higher BAX levels have a longer survival compared with those with reduced BAX expression [Kitada et al. 1998; Pepper et al. 2008]. Anecdotally, patients with CLL who have a specific polymorphism in the BAX gene that reduces its expression also have a shorter survival [Starczynski et al. 2005].
Rationale and development of anti-B-cell lymphoma 2 strategies
Since avoiding apoptosis is a key oncogenic event in lymphoid malignancies, and this is mostly mediated by overexpression of anti-apoptotic molecules such as BCL2 and MCL1, it then makes sense to inhibit them in patients with lymphoid malignancies.
The first attempt to block BCL2 function was oblimersen, an antisense oligonucleotide that is able to reduce the level of BCL2 synthesis both in vitro and in vivo. The drug was evaluated in several clinical trials, including phase III [O’Brien et al. 2009a], but its clinical efficacy was not sufficient for US Food and Drug Administration (FDA) approval and its future remains unclear.
A different strategy, on the other hand, led to the development of BH3 mimetics, a family of agents that holds great promise for the treatment of CLL and other lymphoid malignancies. In CLL cells, excess of BCL2 and other anti-apoptotic proteins sequesters pro-apoptotic molecules such as BIM and others, thus preventing the activation of BAK or BAX and subsequent apoptosis. BH3 mimetics compete for and release these pro-apoptotic proteins, which become able to activate BAX/BAK, leading to MOMP, cytochrome c release into the cytoplasm, caspase activation and cell death. Initially, the development of these agents was hampered by the difficulty in modeling (and therefore disrupting) protein–protein interactions [Petros et al. 2006]. However, nuclear magnetic resonance-based drug screening, parallel synthesis and structure-based drug design allowed for the development of these compounds [Petros et al. 2006]. Several BH3 mimetics have been tested so far, and the difference between them lies on their specificity for the different anti-apoptotic downstream proteins: (i) obatoclax (GX-15-070) and AT101 inhibit all antiapoptotic proteins; (ii) navitoclax (ABT-263) inhibits BCL2, BCL-XL and BCL-W; (iii) venetoclax (ABT-199) inhibits BCL2 only; (iv) A1155463 inhibits BCL-XL only.
Laboratory results with BH3 mimetics
In vitro results using obatoclax showed that increasing drug concentrations gradually reduce cell survival, and also how this effect could be synergistic with that of fludarabine [Campàs et al. 2006]. Moreover, increasing the concentration of AT-101 also resulted in progressive CLL cell death in vitro, even overcoming the MCL1 overexpression induced by stromal cells [Balakrishnan et al. 2009]. Navitoclax is, on the other hand, a more selective and potent drug, as it binds with high affinity to BCL2, BCL-XL and BCL-W, but not to MCL1. As a result, MCL1 overexpression is associated with drug resistance [Pepper et al. 2008]. The drug kills cells through BAX/BAK activation, but it does not activate them directly. Furthermore, MCL1 overexpression is generally induced by stromal cells through cytokines and cell–cell contact in such a way that cytokine deprivation may sensitize tumor cells to navitoclax [van Delft et al. 2006].
Finally, venetoclax was developed with the idea of targeting BCL2 exclusively. This was not easy at first, because the structure of BCL2 is very similar to that of other anti-apoptotic proteins such as BCL-XL and BCL-W, but the investigators succeeded in identifying a compound with such selectivity [Souers et al. 2013]. In vitro studies in primary CLL cells showed that venetoclax is even more potent than navitoclax, and also in murine models where venetoclax prevented tumor growth compared with the control [Souers et al. 2013]. The reason for targeting BCL2 exclusively (and not BCL-XL, BCL-W and MCL-1), is that BCL-XL is also very important for platelet survival and, indeed, one of the most frequent side-effects of navitoclax is thrombocytopenia. In contrast, and by virtue of inhibiting BCL2 exclusively, venetoclax does not cause thrombocytopenia at all [Souers et al. 2013]. Interestingly, in other circumstances, such as in patients with solid tumors, the limiting factor may be neutropenia and not thrombocytopenia, particularly when combining BH3-mimetics with conventional chemo-therapeutic agents. Since BCL2 is crucial for neutrophil survival, BCL-XL-selective inhibitors, such as A1155463, do not cause neutropenia and are currently explored in vitro [Leverson et al. 2015].
Clinical results with B-cell lymphoma 2 inhibitors
Obatoclax and navitoclax
Several BH3-mimetics have been tested in clinical trials, but results are very scanty for obatoclax and AT101. In particular, obatoclax was tested in 26 patients with CLL and, although there was a clear in vivo upregulation of BAX upon drug exposure, only 1 patient (4% of those treated) achieved a partial response. Moreover, there were some neurological side effects that require further investigation [O’Brien et al. 2009b]. Navitoclax was, on the other hand, significantly more effective. In the phase I trial there was a 50% response rate in patients with CLL, although there was also significant thrombocytopenia that correlated with the drug’s concentration in blood [Wilson et al. 2010]. Of note, there was also a significant reduction in the T-cell counts, but without increased opportunistic infections. In view of its clinical efficacy, navitoclax has also been evaluated in combination with rituximab, both in relapsed/refractory and previously untreated patients with CLL (Table 1).
Results of clinical trials evaluating BCL2 inhibitors in patients with chronic lymphocytic leukemia.

This figure includes patients with follicular lymphoma, diffuse large B-cell lymphoma, etc.
17p-, 17p deletion; CRR, complete response rate; FL, frontline; ibru, ibrutinib; idela, idelalisib; m, months; NA, not available; NR, not reached; ORR, overall response rate; OS, overall survival; PFS, progression-free survival; R, rituximab; RR, relapsed/refractory.
Both trials confirmed that navitoclax was effective in CLL and had a manageable safety profile that included neutropenia, thrombocytopenia and transaminitis [Roberts et al. 2015; Kipps et al. 2015]. The toxic effect on T-cells was also confirmed but, by the time the second trial was recruiting, venetoclax was already available. Since thrombocytopenia was a limiting factor in patients with CLL treated with navitoclax, and venetoclax was devoid of this toxicity, further development was subsequently focused on venetoclax [Kipps et al. 2015].
Venetoclax
The first clinical trial of venetoclax in patients with relapsed/refractory CLL revealed impressive results, with a 79% overall response rate (ORR), including complete responses (CR) in 20% of patients [Roberts et al. 2016] (Table 1). Of note, minimal residual disease (MRD) negativity was achieved in 5% of patients, a phenomenon rarely seen in patients with relapsed/refractory disease. Moreover, the results were equally impressive in patients with high-risk disease such as those refractory to fludarabine or with 17p deletion. The median progression-free survival (PFS) had not been reached at 18 months of follow up. In terms of toxicity, neutropenia remained the major side effect but, unfortunately, there were a few cases of severe tumor lysis syndrome (TLS), one of them fatal [Roberts et al. 2016]. This fatal event led to a strict TLS prophylaxis policy (Table 2) together with an adjustment of the dosing schema for the drug. In the amended ramp-up schema the starting dose was 20 mg instead of 50 mg daily, with a gradual increase over 5 weeks up to the maximum dose of 400 mg daily (Figure 3). To date, no further cases of fatal TLS have been observed in this study.
Recommended TLS prophylaxis based on tumor burden (consider all patient co-morbidities before final determination of prophylaxis and monitoring schedule).

ALC, absolute lymphocyte count; LN, lymph node; TLS,
Administer intravenous hydration for any patient who cannot tolerate oral hydration.
Start allopurinol or xanthine oxidase inhibitor 2–3 days prior to initiation of venetoclax.
Evaluate blood chemistries (potassium, uric acid, phosphorus, calcium, and creatinine); review in real time.
For patients at risk of TLS, monitor blood chemistries at 6–8 hours and at 24 hours at each subsequent ramp-up dose.

Venetoclax original and modified administration schedule. The upper panel shows the venetoclax administration schedule in the original dose-escalation cohort. On day −7 (7 days before week 1), an initial single 50 mg dose was administered to 50 patients and a 20 mg dose to 3 patients. Daily administration of a 50 mg dose started in week 1 and was increased to 100–400 mg in week 2. By week 3, patients had reached the designated group dose (150–1200 mg per day). The lower panel shows the modified administration schedule for the 60 patients in the expansion cohort. Daily administration started with 20 mg per day, followed by weekly ramp-up in three steps to 400 mg.
Venetoclax has also been combined with rituximab in patients with relapsed/refractory CLL. The results were also significant with an 86% complete response rate, including CR in 41% of patients (Table 1). Of note, these responses were very deep, with 53% of patients achieving a negative MRD in the bone marrow, which translated into a very long duration of response. Once again, neutropenia emerged as the most important grade 3–4 adverse event, and was observed in half of the patients, although with only 12% cases of febrile neutropenia [Ma et al. 2015]. Another recent study evaluated the efficacy of venetoclax monotherapy in patients with CLL and 17p deletion. Over 100 patients were recruited in this multinational trial and, once again, responses were observed in almost 80% of patients with 40% MRD negativity. Neutropenia was the most frequent grade 3 or 4 side effect, and there were no cases of clinical TLS [Stilgenbauer et al. 2016].
Finally, venetoclax has also been tested in patients failing BTK or PI3K inhibitors, a subgroup of patients with dismal prognosis. The series is much smaller and responses were also less frequent, around 50% with no complete remissions. Grade 3–4 neutropenia and anemia were also relatively common in this patient population [Jones et al. 2015].
Rationale for combining B-cell lymphoma 2 antagonists with Bruton’s tyrosine kinase/phosphoinositide 3’-kinase inhibitors
It is clear that the treatment paradigm for CLL is changing with the advent of novel agents, namely monoclonal antibodies, BTK inhibitors, PI3K inhibitors and BCL2 antagonists (Awan & Byrd, 2014). Unfortunately, none of these agents appears curative and chronic exposure to them has eventually resulted in drug resistance in a proportion of patients. For instance, tumor cells from a proportion of CLL patients chronically exposed to ibrutinib have developed BTK or phospholipase C gamma-2 (PLCγ2) mutations that render the drug ineffective [Woyach et al. 2014b]. Moreover, the same phenomenon has been replicated experimentally upon continuous exposure to venetoclax. At least two different missense mutations within the BCL2 BH3 domain and one missense mutation located in the C-terminal transmembrane domain of BAX have been identified [Fresquet et al. 2014]. As expected, all mutations induced tumor resistance to venetoclax and, what is more worrying, the BAX mutation was also associated with cross-resistance to other neoplastic agents [Fresquet et al. 2014].
It makes sense, then, to test drug combinations in future clinical trials, but one of the challenges ahead of us is how to combine them rationally. Several in vitro and in vivo analyses may be useful in this regard. For instance, ibrutinib (a BTK inhibitor) downregulates anti-apoptotic proteins such as MCL1 and BCL-XL and could be a very good partner for venetoclax. As such, pharmacological profiling has confirmed that this combination results in enhanced cytotoxicity compared with other combinations [Cervantes-Gomez et al. 2015]. Moreover, tumor cells chronically exposed to venetoclax may become resistant to the drug through a variety of mechanisms, and not just the mutations previously mentioned. One of these mechanisms is AKT constitutive activation coupled with upregulation of MCL1 and BCL-XL [Choudhary et al. 2015]. Interestingly, these cells can by resensitized to venetoclax if combined with idelalisib (PI3K inhibitor) or even dual AKT/mTOR inhibitors [Choudhary et al. 2015], or they can be sensitive to a combination of PI3K inhibitors and cyclin-dependent kinase inhibitors [Chiron et al. 2014]. No doubt that clinical trials combining these and other promising agents will clarify this matter in the future.
Conclusion
Resistance to apoptosis is one of the hallmarks of cancer and a particularly important oncogenic event in CLL. This phenomenon is mediated by BCL2/MCL1 overexpression and is associated with resistance to chemotherapy. Several drugs, particularly BH3 mimetics, have been recently developed to target BCL2 and other antiapoptotic proteins. Results with these agents, particularly venetoclax, have been impressive in CLL, including MRD eradication in a sizable proportion of patients. As such, the drug was recently approved (April 2016) by the FDA as monotherapy for patients with CLL and 17p deletion. The toxicity profile was favorable, with neutropenia being the most important side effect. Furthermore, venetoclax appears effective in patients intolerant or refractory to BTK and PI3K inhibitors, and several laboratory studies suggest that BCL2 inhibition could be enhanced by concomitant BTK or PI3K inhibition, providing a rationale for clinical trials in combination.
Footnotes
Funding
This research received no specific grant from any funding agency in the public, commercial, or not-for-profit sectors.
Conflict of interest statement
JD has received lecturing fees from Janssen, Gilead, Roche and Abbvie; research grants from Roche; and consulting fees from Janssen, Gilead, Roche and Abbvie. VOM and PM declare no conflicts of interest.