
Abstract
There have been major advances in our understanding of the multiple interactions between malignant cells and the innate and adaptive immune system. While the attention of immunologists has hitherto focused on solid tumors, the specific immunobiology of acute leukemias is now becoming defined. These discoveries have pointed the way to immune interventions building on the established graft-versus-leukemia (GVL) effect from hematopoietic stem-cell transplant (HSCT) and extending immunotherapy beyond HSCT to individuals with acute leukemia with a diversity of immune manipulations early in the course of the leukemia. At present, clinical results are in their infancy. In the coming years larger studies will better define the place of immunotherapy in the management of acute leukemias and lead to treatment approaches that combine conventional chemotherapy, immunotherapy and HSCT to achieve durable cures.
Introduction
One of the major obstacles to the cure of acute leukemia (AL) is its propensity to relapse after chemotherapy or hematopoietic stem-cell transplantation (HSCT). Treatments exploiting immune responses against malignant disease and more recently leukemia have advanced in recent years. The rationale for immunotherapy in AL is supported by evidence for immune surveillance in the development of leukemia. Notably, solid organ transplant recipients on lifelong immunosuppression have an increased risk of developing AL [Adami et al. 2003; Vajdic et al. 2006; Villeneuve et al. 2007; Gale and Opelz, 2012]. Furthermore, swift lymphocyte reconstitution after chemotherapy is associated with improved relapse-free survival [Ohnishi et al. 1998; Behl et al. 2006; De Angulo et al. 2008]. In addition to these indirect observations, the efficacy of allogeneic HSCT provides clear evidence that donor-derived immune system can attack the recipient’s leukemic cells. This graft-versus-leukemia (GVL) effect is mediated by donor T cells recognizing alloantigens and leukemia-associated antigens presented by the leukemia and by natural killer (NK) cells through killer immunoglobulin-like receptor (KIR) and KIR–ligand interaction [Barrett, 2008]. Escape of leukemia in the form of relapse after either allogeneic HSCT or from some form of autologous immune control following induction chemotherapy control is, however, all too frequent. Some of the immune evasion mechanisms discovered in solid tumors are shared by AL, but the field is less well explored in hematological malignancies. Better understanding of these mechanisms is essential for the design of rational and effective immunotherapy for AL.
The first section of this review addresses the mechanisms by which AL evades immune control. In the second section, current developments and recent clinical experience with immunotherapy, designed both to overcome immune evasion and to deliver lethal damage to the leukemia cell, are examined. Finally, we discuss how immunotherapy can be incorporated into transplant and nontransplant treatment approaches for AL.
How leukemia evades the immune system
Recent studies have revealed a broad range of strategies employed by leukemia to block cytotoxicity of T cells and NK cells against leukemia. Most research has involved myeloid malignancies and less is known about acute lymphoblastic leukemia (ALL). In the process of disabling immune attack, leukemia causes many changes in T and NK cell phenotype and function. This process is termed immune editing. Interactions of leukemia with the innate and adaptive immune system are diverse. They are described below and summarized in Figure 1.

Immune evasion mechanisms seen in acute leukemia.
Lymphocyte phenotype changes due to immune editing
T cells in acute myeloblastic leukemia (AML) show many abnormalities. The spectrum of circulating T cells from the most recent thymic emigrants (RTE) to the end effector cells is perturbed with a reduction of RTE [Li et al. 2009] and an excess of CD57+ CD45RO + end effectors [A.J.B. personal observation]. There is a general increase in the expression of ‘exhaustion markers’ such as programmed death 1 (PD1) and T-cell immunoglobulin domain and mucin domain 3 (TIM3) in T cells in AML [Zhou et al. 2011]. These markers characterize the end effector stage of T-cell evolution following antigen stimulation and expansion and can imply T-cell senescence. T cells from AML patients have aberrant T-cell activation patterns based on gene expression profiling and they fail to synapse normally with their leukemic targets [Le Dieu et al. 2009]. There is an increased proportion of regulatory T cells (Tregs) in both AML and ALL [Szczepanski et al. 2009; Kanakry et al. 2011; Shenghui et al. 2011] which persists into remission. Thus leukemia-induced T-cell tolerance coupled with a preponderance of Tregs reduces the ability of cytotoxic T cells to target leukemia.
NK cells also show significant phenotypic changes with a tendency for overexpression of surface molecules that reduce NK cytotoxicity (i.e. inhibitory receptors) such as NK group 2, member 2A (NKG2A) which recognizes human leukocyte antigen (HLA) E expressed by the leukemia and downregulation of activating receptors such as NKG2D [Davies et al. 2014]. Overnight co-incubation of NK cells from healthy donors with AML blasts induces NKG2A and inhibits their cytotoxic capacity, demonstrating that AML blasts directly impair innate immune function [Stringaris et al. 2014].
Tregs
Tregs, characterized by expression of the transcription factor FoxP3 [Sakaguchi et al. 2010], suppress effector T cells. Tregs can diminish anticancer T-cell immunity [Zou, 2006] and are increased in both murine and human AML [Wang et al. 2005; Szczepanski et al. 2009; Zhou et al. 2009; Shenghui et al. 2011]. Treg numbers at presentation of leukemia are associated with poor response to chemotherapy [Szczepanski et al. 2009; Shenghui et al. 2011]. Presence of Tregs can limit effective immunotherapy, leading to attempts to block Tregs prior to, or in conjunction with, various other immunotherapeutic modalities (discussed in detail below).
Myeloid-derived suppressor cells (MDSC)
A heterogeneous population of immature myeloid-derived cells expands in pathological conditions including cancer [Gabrilovich and Nagaraj, 2009]. Granulocytic MDSC (CD11b + CD33 + CD14-) and monocytic MDSC (CD11b+ CD33+ CD14+) are the two major subtypes described in leukemia [Abu Alex et al. 2012; Christiansson et al. 2013; Jitschin et al. 2014]. MDSCs suppress T-cell responses through various mechanisms including
The role of MDSCs in suppressing the proliferation and function of leukemia antigen-specific T cells is insufficiently understood. However, their contribution is suggested by recent studies where we found that both CD34+ and CD33+ AML blasts suppress T-cell proliferation in an MDSC-like manner [Ito et al. 2013]. Furthermore, dendritic cells (DCs) grown from AML blasts are functionally impaired and induce T-cell anergy [Mohty et al. 2001; Narita et al. 2001].
Establishment of a milieu hostile to immune cells
Various defects observed in T cell and NK cell function can be caused by soluble factors secreted by leukemic cells [Orleans-Lindsay et al. 2001]. Some leukemic cells have abnormal enzymatic activities, causing depletion of substrates or excess of metabolites, both of which can lead to immunosuppression. A key transcription factors involved in tumorigenesis, also constitutively expressed in myeloid leukemia is signal transducer and activator of transcription 3 (STAT3) [Yu et al. 2009]. STAT3 targets genes that stimulate cell proliferation, prevent apoptosis, promote angiogenesis and facilitate tumor immune evasion. It plays a significant role in subversion of host immune responses, causing accumulation and activation of immunosuppressive cells [Tregs, T helper 17 (Th17) cells and MDSC] while reducing DC function [Rebe et al. 2013]. In a murine model of AML, STAT3 blockade was shown to rescue the immune response to AML, leading to remission by upregulation of major histocompatibility complex (MHC) class II, costimulatory and proinflammatory mediators, while coinhibitory PD-L1, programmed death–ligand 1 (PD-L1) molecule was downregulated [Hossain et al. 2014].
IDO
IDO is a rate-limiting enzyme in tryptophan degradation into kynurenine [Taylor and Feng, 1991; Mellor and Munn, 1999]. It is constitutively expressed in many human tumors and contributes to cancer immune evasion [Uyttenhove et al. 2003]. In AML, IDO expression is described in about half of patients at diagnosis [Curti et al. 2007a] and IDO enzymatic activity is elevated [Corm et al. 2009]. IDO depletes tryptophan, which leads to inhibition of T cell and NK cell proliferation [Mellor and Munn, 1999; Frumento et al. 2002]. Furthermore, IDO-expressing AML cells induce Tregs [Curti et al. 2007b, 2010] and patients with IDO-expressing AML have higher circulating Treg frequencies [Curti et al. 2007b]. Importantly, IDO expression level and activity in AML is associated with poor clinical outcomes [Chamuleau et al. 2008; Corm et al. 2009].
l -Arginine, nitric oxide synthase (NOS) and arginase
Leukemia evasion from immune attack
It is well described that loss or downregulation of MHC class I molecules by cancer cells make them invisible to cytotoxic T cells [Khong and Restifo, 2002]. After haploidentical HSCT for AML, loss of mismatched HLA haplotype due to uniparental disomy of chromosome 6p was reported as one of the mechanisms of post-transplant relapse [Vago et al. 2009; Crucitti et al. 2015]. Subsequently, partial loss of HLA was demonstrated and implicated as the cause of post-transplant leukemic relapse in matched related [Stolzel et al. 2012] and unrelated donor HSCT [Toffalori et al. 2012]. Factors that affect the ability of HLA molecules to present antigen to T lymphocytes also matter: class II associated invariant chain peptide (CLIP) occupies the HLA–class II antigen groove and must be released before antigenic peptides can bind to the groove [Romagnoli and Germain, 1994; Sloan et al. 1995]. High expression of CLIP associated with failure of the leukemia antigen to occupy the antigen presentation groove is associated with worse clinical outcome in AML, implying a failure of antigen presentation by the leukemia [van Luijn et al. 2010]. Loss of costimulation molecules on leukemia blasts can cause T-cell anergy. Leukemia blasts commonly lack CD80 expression while CD86 expression level can be heterogeneous [Whiteway et al. 2003; Graf et al. 2005; Mansour et al. 2014]. Diminished expression of costimulatory molecules on AML blasts [Vollmer et al. 2003] may be associated with worse clinical outcome [Whiteway et al. 2003].
Abnormal cells, such as virus infected cells and malignant cells, lack MHC class I expression, allowing NK cells to exert their cytotoxic function by recognition of ‘missing-self’ (by lack of interaction between cognate HLA molecule and KIR). In the HLA mismatched transplant setting, donor NK cells and their KIRs recognize disparity in the MHC class molecules expressed on leukemic blasts and exert a GVL effect [Ruggeri et al. 1999]. Underexpression of MHC class I molecules in mismatched HSCT setting should render the leukemia susceptible to allogeneic NK-cell cytotoxicity. However, leukemias also escape NK-cell mediated lysis by various mechanisms. The activating NK cell receptor, NKG2D, recognizes the major immunogene complex (MIC) A/B expressed on leukemia blasts [Bauer et al. 1999]. MIC A/B is overexpressed by myeloid leukemias but is also shed, blocking NK cell engagement with the target [Sconocchia et al. 2005]. AML blasts with high expression of inhibitory KIR ligands, HLA-Bw4 and C, are less susceptible to NK-cell mediated lysis [Demanet et al. 2004; Verheyden et al. 2009]. HLA-G, one of ligands for KIR2DL4 [Hofmeister and Weiss, 2003], is also expressed by AML blasts and is implicated in the inhibition of NK-cell function [Yan et al. 2008]. NK cell function is also blocked through interaction between glucocorticoid-induced tumor necrosis factor-related protein (GITR) on the NK cell and its ligand, GITRL, expressed or secreted by AML cells [Baessler et al. 2009].
Promotion of immune checkpoints
Immune checkpoint pathways are important for control of T-cell activation and maintenance of selftolerance [Fife and Bluestone, 2008]. Various cancers use immune checkpoints to their survival advantage [Pardoll, 2012]. Cytotoxic T-lymphocyte associated antigen 4 (CTLA-4) is expressed on T cells and shares the same ligands, CD80 and CD86, with CD28. Ligation of CD28 to these ligands elicits costimulatory signaling and T-cell activation. However, when CTLA-4 binds to CD80 or CD86, early T-cell activation is inhibited [Schwartz, 1992; Fife and Bluestone, 2008; Pardoll, 2012]. Furthermore, CTLA-4 increases inhibitory cytokine production by CD4+ T cells [Chen et al. 1998]. CTLA-4 is expressed in up to 85% of leukemia blasts [Pistillo et al. 2003] and its blockade may have therapeutic benefit by increasing the activity and numbers of AML-reactive T cells [Zhong et al. 2006].
PD-1 is expressed on antigen-experienced and activated T cells, B cells and NK cells [Fife and Bluestone, 2008]. There are two known ligands of PD-1, PD-L1 and PD-L2: PD-L1 is broadly expressed on peripheral tissues while PDL2 is expressed in a more limited manner on DCs and monocytes [Fife and Bluestone, 2008]. The PD-1 pathway induces peripheral T-cell tolerance by directly inhibiting activated T cells and by enhancing Treg-mediated immune suppression [Francisco et al. 2010]. PD-L1 is upregulated on various cancers [Dong et al. 2002; Pardoll, 2012]. Between a fifth to a half of AL express PD-L1 [Salih et al. 2006; Chen et al. 2008; Berthon et al. 2010]. Enhanced PD-1 expression on tumor-infiltrating lymphocytes (TILs) or circulating peripheral blood T cells is a well-described phenomenon in various human cancers, and PD-1 expression on T cells may be increased in patients with AL [Ahmadzadeh et al. 2009; Sfanos et al. 2009; Christiansson et al. 2013; Yang et al. 2014]. In murine leukemia models, PD-1 expression on cytotoxic T cells increased with AML progression [Zhou et al. 2010; Zhou et al. 2011] and in the post-transplant setting, causing T-cell exhaustion [Asakura et al. 2010; Flutter et al. 2010].
Advances in the field of immunotherapy for AL
The goals for immunotherapy (excluding antibody treatment which is not discussed in this review) in the treatment and cure of AL are threefold: (1) to boost cytotoxicity of lymphocytes targeting the leukemia; (2) to block the process of immune editing and recreate a permissive environment for T cell and NK cell function; and (3) to integrate immunotherapy with conventional existing approaches of chemotherapy and HSCT. Various immunotherapeutic approaches for AL are summarized in Figure 2.
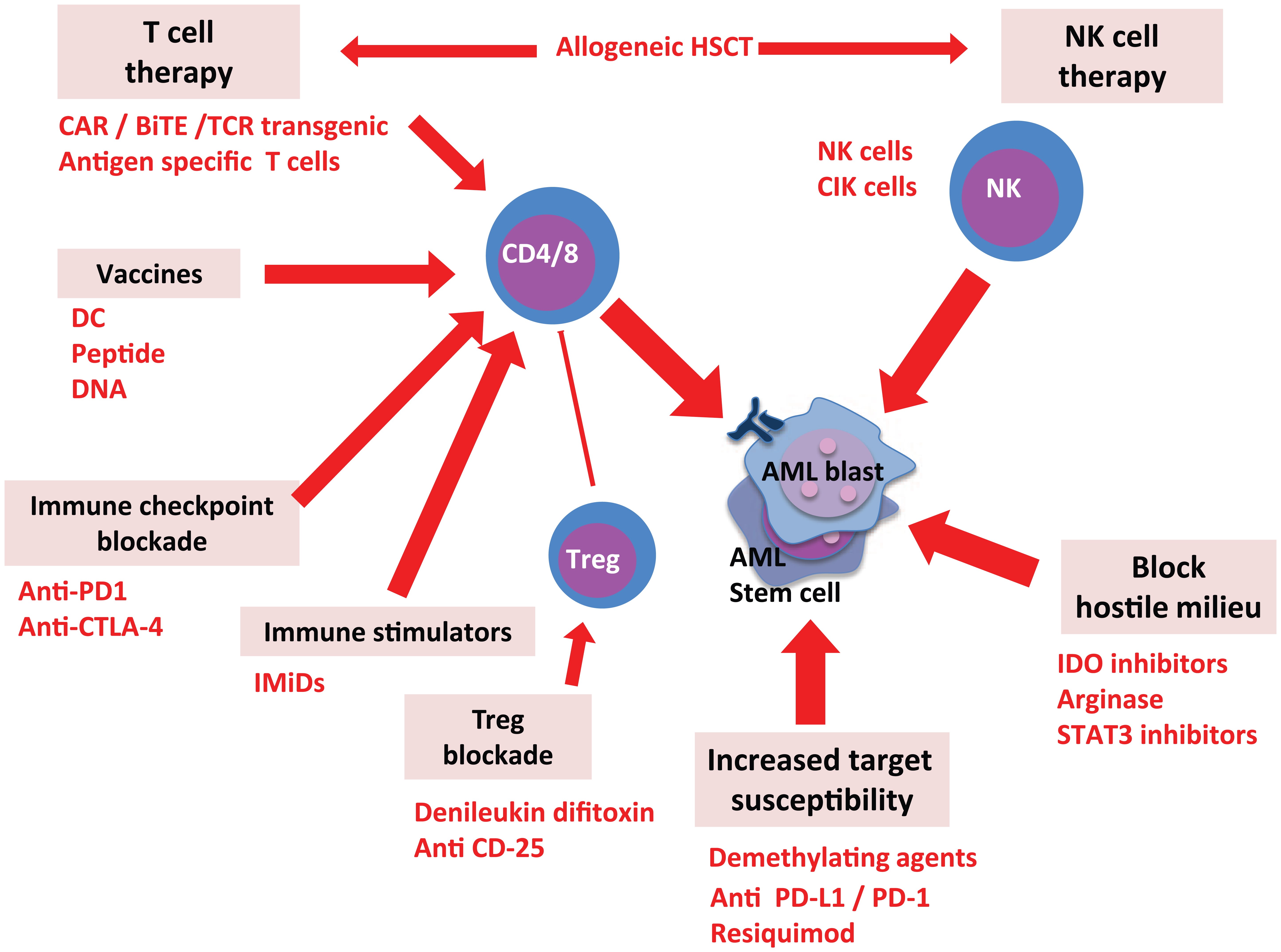
Exploiting therapeutic effects by targeting leukemia’s immune evasion mechanisms.
Allogeneic HSCT
Immunotherapy for AL involves two distinct therapeutic domains: allogeneic HSCT and nontransplant treatments. Immunotherapy approaches are often first explored in patients who have received allogeneic HSCT either to prevent or treat relapsing leukemia. While many immunotherapy strategies apply in or outside the context of HSCT, the alloimmune GVL effect is both unique and powerful, and is discussed separately here.
Allogeneic HSCT is still the only undisputed immune-based approach to curing leukemias resistant to chemotherapy alone. The GVL effect is mediated by both T cells and NK cells [Bleakley and Riddell, 2004; Barrett, 2008; Warren and Deeg, 2013]. While the stem-cell source (bone marrow, peripheral blood or cord blood), degree of compatibility (identical twin, matched related, matched unrelated and haplotype matched related donors) and transplant approach [conditioning regimen and method of graft-versus-host disease (GVHD) prevention] can all influence outcome, the major factor determining the survival and cure of AL is the status of the leukemia at the time of transplant.
Patients of standard risk with AL transplanted in first remission have a relapse rate of 20% or less; those in second or subsequent remission have an intermediate relapse risk of around 40%, while patients transplanted with overt disease either because of refractoriness to induction treatment or uncontrolled relapse had a relapse risk around 60% [Barrett, 2008). Despite numerous strategies manipulating the transplant schedule to decrease relapse rates, these statistics have not changed significantly for over 40 years. This is in part due to the fact that approaches that boost the GVL effect are constrained by the increase in morbidity and mortality from GVHD, and increased ablation of leukemia by radiation and chemotherapy is limited by increased mortality from nonrelapse causes. In addition, despite many innovative attempts to control relapsed disease by boosting GVL, relapse of AL after HSCT is largely incurable and still carries a dismal prognosis. Palliative treatment aside, the standard treatment of relapsed AL is chemotherapy followed by donor lymphocytes infusion (DLI). While up to 20% of patients may enjoy prolonged survival, few are cured and relapse occurring within 6 months of transplant is almost universally incurable [Treleaven and Barrett, 2009].
DLI
The full realization that GVL was a major contributor to the curative effect of allogeneic HSCT in the 1980s led logically to the use of DLI to prevent or treat relapse after transplant. Unfortunately, the first demonstrations that DLI could achieve durable remissions in relapsed CML were not borne out in AL and myelodysplastic syndromes (MDS) where the therapeutic benefit of DLI is at best modest. The failure of DLI to fulfill its promising results with AL stimulated a number of approaches to improve DLI as detailed in Table 1. Treg-depleted DLI has been used to augment GVL [ClinicalTrials.gov identifier: NCT00675831, NCT00987987]. Manipulation of Tregs in the context of HSCT (both in the graft and in the host after transplantation) has two implications: depletion of Tregs may enhance GVL, but enrichment of Tregs is efficacious for prevention and treatment of GVHD [Martelli et al. 2014]. Further studies are required to identify the optimal therapeutic approach involving Treg-mediated pathways to achieve a fine balance between GVL and GVHD. Unfortunately there is insufficient data from any strategy to determine a definitive role for ‘augmented’ DLI. Only the prophylactic application of DLI has any clear advantage.
Strategies used to improve therapeutic efficacies of DLI.

AML, acute myeloblastic leukemia; CTLA-4, cytotoxic T lymphocyte antigen-4; DLI, donor lymphocytes infusion; GM-CSF, granulocyte-macrophage colony-stimulating factor; GVHD, graft-versus-host disease; IFN, interferon; IL, interleukin; MDS, myelodysplastic syndromes; PD1, programmed death 1; PD-L1, PDL1, programmed death–ligand 1; Treg, T-cell regulator
Immunotherapy beyond HSCT
New developments in immunological understanding and ability to manipulate immune cells are beginning to break the impasse of GVL linked to GVHD and improve the curative potential of allogeneic HSCT. Relapse after HSCT with its extremely poor prognosis has become a proving ground for the most innovative and experimental immunotherapy, appropriate for such desperate circumstances. Thus many of the strategies described below were initially developed or have only been applied in the context of HSCT. However, the application of novel immunotherapy outside the context of HSCT is now gaining momentum. These new strategies can be categorized as: (1) lymphocyte products to deliver enhanced and specific anti-leukemic cytotoxic (cell therapy); (2) vaccines; and (3) treatments to boost immunity and enhance leukemia’s susceptibility to immune system.
Cell therapy
Chimeric antigen receptor modified T cells (CAR-T). The remarkable therapeutic successes using CAR-modified T cells to treat leukemia have attracted wide enthusiasm for CAR cell therapy and T-cell therapy in general. The principle is to insert a T-cell receptor (TCR)/costimulatory molecule/linker/antigen binding domain derived from the fusion of the variable regions of the heavy and light chains of immunoglobulins specific for a leukemia surface antigen (e.g. anti CD19) construct into T cells. The antibody binds surface molecules on the leukemia, triggers TCR activation and directs T-cell cytotoxicity to the leukemia.
Initial reports of remissions achieved in ALL and chronic lymphocytic leukemia (CLL) by Porter and colleagues [Porter et al. 2011; Grupp et al. 2013] have led many investigators to adopt and further refine this technology. Key developments have been the improvement of the construct with substitution of 41BB for CD28 as the costimulatory molecule, and use of long-lived viral specific T cells rather than CD3/CD28 bead stimulated T cells as the vehicle for TCR transduction generating CAR cells to improve persistence. Other approaches under investigation employ virus-free transduction systems (sleeping beauty transposons) and the development of NK CAR cells as an alternative to T cells. The most advanced clinical trials with CAR cells in AL have been in patients with ALL. Maude and colleagues described the treatment of 30 adults and children with relapsed or refractory ALL with autologous T cells transduced with a CD19-directed CAR (CTL019) [Maude et al. 2014]. All the patients had a cytokine-release syndrome which was severe and required anti-IL-6 treatment in about a quarter of the recipients. A total of 27 achieved a complete remission, with a 6 month event-free survival and overall survival rate of 67% and 78%, respectively. The CAR cells persisted and were associated with a prolonged B-cell aplasia. These results were all the more remarkable because durable remissions were seen in patients failing blinatumomab and HSCT.
There has been considerable interest in using CAR technology to target myeloid leukemias. CD123 is overexpressed by AML but it is also well-expressed on normal myeloid cells, although expression on hematopoietic stem and progenitor cells is weaker. Nevertheless the risk of marrow ablation limits the application of antimyeloid leukemia CAR cells to pretransplant conditioning where healthy stem cells can be infused after ablation of the CAR cells.
While CAR cells have alerted the oncology community to the potency of T-cell-based therapy and stimulated considerable interest of pharmaceutical companies in developing cell therapy, they have some important limitations. Firstly their use in leukemia is restricted by the lack of antigens that are uniquely expressed by the leukemic cell and not the normal counterpart. A further problem is the ability of the leukemia to downregulate surface expression of molecules such as CD19 – a common form of immune evasion in B-cell malignancies. It may, in future, be possible to target the much larger population of intracellular tumor specific antigens recognized by T cells when presented as peptide fragments through the tumor MHC, by developing antibodies specific for the antigenic-peptide MHC complex. Such antibodies have been developed for WT1 and PR1, but their application would be restricted to the specific HLA molecule expressing the tumor peptide (usually HLA A2) [Alatrash et al. 2012; Veomett et al. 2014].
TCR transduction. Several investigators have used T cells genetically modified to express a TCR specific for a single MHC class I or II expressed antigen. Preclinical studies suggest efficacy and clinical trials are in progress [Xue et al. 2010; Katsuhara et al. 2015]
Bispecific T-cell engagers (BiTEs). Bispecific antibodies target both a tumor antigen and CD3. When the molecule targets the tumor, the CD3 binds and activates circulating T cells at the site of the tumor [Huehls et al. 2015]. Blinatumomab, a BiTE which targets CD19 and CD3, shows important antitumor activity in patients with B-cell malignancies. The US Food and Drug Administration (FDA) gave accelerated approval for blinatumomab to be used in treatment of ALL. Early experience with blinatumomab in ALL is promising [Topp et al. 2011]. The agent eliminated minimal residual disease in 16/20 patients with adult ALL and prolonged remission [Topp et al. 2012]. Blinatumomab is also active for primary refractory or relapsed pre-B ALL with response [complete remission (CR) or complete response with partial hematologic recovery (CRh)] rate of over 40% [Topp et al. 2015]. However, similar to CAR cells, the lack of a specific myeloid leukemia surface antigen specific for AML currently limits the BiTE strategy to B-cell ALL.
Antigen specific T cells. ALs express a variety of leukemia-associated antigens (LAA), including classical tumor antigens, via their MHC class I and II molecules, rendering them susceptible to attack by tumor antigen-specific T cells [Goswami et al. 2014]. It is important to note that such CTL can target leukemia progenitors, which increases their likelihood of therapeutic efficiency [Quintarelli et al. 2011; Schneider et al. 2015]. ALs commonly express multiple LAA, making it possible to target them with T cells recognizing more than one antigen, thereby reducing the risk of tumor escape by antigenic downregulation. We have shown that multi-LAA-specific cytotoxic CD4 and CD8 T cells can be generated against peptide mixtures of tumor antigens in 2–3 week expansion cultures using T cells from healthy donors or leukemia patients in remission [Weber et al. 2013a, 2013b]. WT1 specific LAA specific T cells generated using peptide antigens have been shown to have efficacy in AML [Weber et al. 2013b]. In HSCT, the opportunity to target minor histocompatibility and LAA antigens with donor T cells has been developed in several centers and early clinical trials show promise in patients relapsing after HSCT with AL. In a study from Seattle, 11 high risk patients after HSCT received donor-derived HLA-A2 restricted CD8+ T-cell clones specific for the WT1126–134 peptide [Chapuis et al. 2013]. No GVHD occurred. The study highlighted the need to expand T cells in vitro in the presence of IL-21: patients who received T cells expanded with IL-21 were alive and achieved durable remission after infusion of CTL, while most patients receiving CTL without IL-21 expansion had short CTL persistence and relapsed quickly [Chapuis et al. 2013].
A phase I clinical trial at the Memorial Sloan Kettering Cancer Institute gave WT-1 specific T cells to allogeneic HSCT recipients with AML, ALL or MDS. WT-1 specific T cells were generated by DCs pulsed with pools of overlapping peptides spanning the WT1 protein. Minimal residual disease was transiently reduced after T-cell infusion without causing GVHD [O’Reilly et al. 2010]. Other investigators have exploited minor histocompatibility antigen (mHA) differences between donor and patient to generate mHA-specific T cells to target leukemia following HSCT [Bleakley and Riddell, 2011].
NK cell therapy. It is now feasible to expand NK cells ex vivo for cell therapy. NK cells can be selected with magnetic beads coated with CD56 from an apheresis collection and can be expanded with Epstein–Barr virus (EBV) transformed B cell lines or K562 cell lines expressing costimulatory molecules and membrane-bound IL-15 or IL-21 [Denman et al. 2012; Shah et al. 2013]. Early trials show that NK cell infusions are without the risk of GVHD. Two recent uncontrolled studies suggest that NK cells may be efficacious in preventing relapse or treating refractory AML [Miller et al. 2005; Rubnitz et al. 2010]. After lymphodepleting treatment with cyclophosphamide and fludarabine, haploidentical NK cells were administered to children with AML in first complete remission. The 2 year event-free survival was 100% [Rubnitz et al. 2010]. A similar lymphodepletion/haploidentical NK cell infusion study in 19 adults with refractory AML achieved 5 complete remissions [Miller et al. 2005]. These results are preliminary and need further validation.
Cytokine induced killer (CIK) cells. CIK cells represent a population of T cells and NK cells with cytotoxic capacity induced by IL-2 and other cytokines. While numerous studies have explored these cells manufactured in a variety of ways, their overall impact on control of AL has been disappointing. Studies with CIK cells are well reviewed by Pittari and colleagues [Pittari et al. 2015].
Vaccines
Immunotherapy of AL experienced a brief period of enthusiasm in the 1970s when several groups explored vaccination of AL patients in remission with irradiated leukemia cells and Bacille Calmette Guerin (BCG) vaccine given either together or separately. These studies were inconclusive and were soon bypassed by the development of HSCT as a curative approach. More recently, with better knowledge of antigen presentation and leukemia antigens, and the ability to monitor residual disease and immune responses, there is renewed enthusiasm for vaccination to prevent relapse after remission induction or after HSCT. Vaccination approaches are diverse. They may target specific leukemia antigens such as WT1 and BCR-ABL, or undefined antigens derived from entire leukemia cells lysed or fused with antigen-presenting cells (APC), or leukemia lines engineered to secrete granulocyte-macrophage colony-stimulating factor (GM-CSF) [Zeng et al. 2003; Rosenblatt and Avigan, 2006; Ho et al. 2009]. Vaccines can be delivered as APC (DCs or leukemia cells) [Palucka and Banchereau, 2013; Schurch et al. 2013] or as proteins, peptides or DNA [Rice et al. 2008; Tabarkiewicz and Giannopoulos, 2010; Rezvani, 2011].
Recent developments on leukemia vaccines in AL. Single epitopes of WT1 and PR1 peptides were given with adjuvant to patients with AML and MDS in a phase I/II study. Although there were impressive falls in minimal residual disease (MRD) in association with a rise in PR1 or WT1 specific T cells, the responses were not sustained and overall clinical responses were modest. Nine clinical trials using WT1 peptides in patients with MDS and/or AML, published between 2004 and 2012, were recently reviewed [Di Stasi et al. 2015]. A total of 51 patients received vaccine. Vaccination was safe and associated with clinical responses with a few patients achieving long-term disease-free survival sustained up to 8 years. Responses correlated with the detection of WT1-specific T cells and normalization/reduction of WT1 mRNA levels. Controlled clinical trials are needed before these promising results can be properly assessed.
DC vaccines. Anguille and colleagues have reviewed the strategy of using DCs as antigen presenting cells transduced with tumor antigen DNA [Anguille et al. 2014]. This group described encouraging results with a WT1-transduced DC in patients with AML: five of 10 patients achieved complete remissions including two patients refractory to chemotherapy [van Tendeloo et al. 2010]. These results are encouraging but have not been reproduced in a larger patient series. A major limitation of vaccination approaches is the fact that most defined leukemia antigens are products of normal genes overexpressed or selectively expressed in leukemia cells. The immune system is finely balanced to distinguish foreign antigens from self antigens. In effect, cancer vaccination aims to break tolerance to self and elicit an ‘autoimmune’ response. Thus one of the major hurdles for effective vaccination is overcoming the central and peripheral tolerance to self antigens.
Vaccines after HSCT. The opportunity provided by HSCT to combine vaccines with the immune cells from a nontolerized donor has led to studies exploring vaccination after HSCT. The association of vaccines with HSCT has some unique advantages. Firstly, the lower tumor burden early after HSCT phase, which is an ideal setting for anti-tumor immune responses to operate. Secondly, the unique immune milieu around the time of the transplantation characterized by lymphopenia, Treg depletion, and the release of growth factors such as IL-7, IL-15, and IL-21, is permissive to the generation of antileukemia immune responses. Thirdly, HSCT provides the opportunity to associate vaccination with administration of leukemia specific cells, either by donor vaccination or vaccinating the patient post cell infusion [Rezvani and Barrett, 2008; Barrett and Rezvani, 2009]. Alternatively vaccine-primed lymphocytes can be collected by apheresis and reinfused after transplant with further vaccination to boost the response. There are also limitations to the efficacy of vaccine given after allogeneic HSCT: antigen-specific CD8+ T cells during this time may be at risk for rapid induction of senescence [Rezvani and Barrett, 2008; Barrett and Rezvani, 2009].
In summary, vaccination approaches have shown promise but, frustratingly in the absence of substantial well-controlled trials in AL, the field remains in its infancy.
Treatments to boost immunity and enhance leukemia’s susceptibility to immune system
Immune checkpoint blockade. PD-L1 blockade on the tumor cell or PD1 blockade targeting the T cell is emerging as one of the most powerful strategies to enhance T-cell effectiveness against cancer [Pardoll, 2012]. Antibodies or blocking molecules have been developed targeting the key checkpoint inhibitors illustrated in Figure 1. Most experience with these agents comes from the field of solid tumor immunotherapy [Brahmer et al. 2012; Prieto et al. 2012; Topalian et al. 2012]. However, anti-CTLA-4 has been used in a preclinical AML model [LaBelle et al. 2002; Zhong et al. 2006] and also for treatment of patients with post-HSCT relapse [Bashey et al. 2009]. This study demonstrated that CTLA-4 inhibition with ipilimumab does not increase the risk of GVHD, even in patients who received DLI in conjunction [Bashey et al. 2009]. While promising responses were achieved, the study was too small to clearly identify efficacy of the agent in AL. Ongoing trials are testing the safety and efficacy of CTLA-4 blockade in both post-transplant relapse settings and nontransplant settings [ClinicalTrials.gov identifier: NCT00060372, NCT01757639, NCT01822509]. Similarly, preclinical data for PD-1/PD-L1 blockade have shown that the target has therapeutic potential in leukemia [Zhang et al. 2009]. PD-1/PD-L1 blockade has been tested and actively investigated in various hematologic malignancies [Armand, 2015] [ClinicalTrials.gov identifier: NCT02117219, NCT01953692, NCT02332980], but clinical experiences in AL remain limited. In a phase I study of anti-PD-1 antibody for hematologic malignancies that enrolled total of 17 patients, 8 patients had AML [Berger et al. 2008]. None of the patients experienced toxicity attributable to the agent. Majority of them did not show any response, except for one patient who demonstrated marked reduction in peripheral blood blasts burden. The result suggests that PD-1/PD-L1 blockade can be effective in a selected setting. Anti-PD-1 antibody along with DC vaccine for AML [ClinicalTrials.gov identifier: NCT01096602] is being tested. While checkpoint inhibition is a promising novel treatment, further studies are required to understand the most efficacious methods of administration to exploit the possible clinical benefit.
Enhancing leukemia’s susceptibility to immune attack. In addition to immune checkpoint inhibition, there are other agents that aim to restore leukemia’s susceptibility to immune surveillance. The mechanisms by which these agents potentially exert therapeutic effect are not completely understood. Many of these agents have been proven to have some efficacy in other hematologic malignancies, such as myelodysplastic syndrome, but clinical data for AL is still limited.
Immunomodulatory drugs (IMiDs). IMiDs include prototypical thalidomide and its structural analogs lenalidomide and pomalidomide [Zeidner and Foster, 2015]. IMiDs have protean immunomodulatory functions that work in concert to reverse various immune evasion mechanisms described in Figure 1.
They modulate cytokine levels, such as suppressing tumour necrosis factor-α (TNF-α) [Moreira et al. 1993; Corral et al. 1999] and augmenting other pro-inflammatory cytokines including IL-2 and interferon (IFN) γ and promote type 1 T cell (Th1) responses [Haslett et al. 1998; Dredge et al. 2002].
They upregulate costimulatory molecules (such as CD80 and CD86) [Aue et al. 2009] and tumor-associated antigens, making malignant cells more readily recognizable to effector T cells [Henry et al. 2013].
They downregulate coinhibitory molecules and overcome immune checkpoint pathway-related immune evasion mechanisms [LeBlanc et al. 2004; Luptakova et al. 2013].
They allow cytotoxic T lymphocytes to form normal immunological synapse with leukemic blasts [Ramsay et al. 2008; Shanafelt et al. 2013].
They inhibit Tregs [Galustian et al. 2009].
They restore NK cell-mediated tumor lysis [Davies et al. 2001; Krieg and Ullrich, 2012; Lagrue et al. 2015].
Epigenetic agents. Another therapeutic approach for AL is to restore normal gene expression by modifying epigenetic changes and remodeling chromatin structure by DNA methyltransferase (DNMT) inhibitors or histone deacetylase (HDAC) inhibitors. AL cells frequently have gene mutations that affect DNA methylation and/or histone post-translational modifications, such as DNA methyltransferase-3A (DNMT3A) [Marcucci et al. 2012], Tet methylcytosine dioxygenase 2 (TET2) [Metzeler et al. 2011; Weissmann et al. 2012], additional sex combs-like 1 (ASXL-1) [Pratcorona et al. 2012], and isocitrate dehydrogenase (IDH) 1 and 2 [Figueroa et al. 2010; Janin et al. 2014].
In addition to restoring normal gene expression pattern and inducing leukemic blasts differentiation (or phenotypic modifications toward maturation), epigenetic agents may have various immunomodulatory functions by either downregulating or upregulating various genes involved in regulation of T-cell proliferation, activity and plasticity [Leoni et al. 2002; Reddy et al. 2004; Tao et al. 2007; Choi et al. 2010; Sanchez-Abarca et al. 2010]. When DNMT inhibitors are used as a single agent (either azacitidine or decitabine) for AML, they confer modest overall response rates of about 20% [Silverman et al. 2006; Cashen et al. 2010; Fenaux et al. 2010; Kantarjian et al. 2012; Lubbert et al. 2012]. Data for single agent efficacy of HDAC inhibitors (such as vorinostat, romidepsin, valproic acid and entinostat) in AML is further limited and less promising, with reported response rates of 0 to <20% in phase I and II trials [Bug et al. 2005; Kuendgen et al. 2005; Raffoux et al. 2005; Gojo et al. 2007; Garcia-Manero et al. 2008; Odenike et al. 2008; Schaefer et al. 2009]. However, combination of DNMT inhibitors and HDAC inhibitors seem to have much better efficacy in AML [Garcia-Manero et al. 2006; Blum et al. 2007; Soriano et al. 2007; Prebet et al. 2014; Issa et al. 2015]. DNMT inhibitors and HDAC inhibitors have been tried in conjunction with IMiDs and combinations are reported to have synergistic effects [Raza et al. 2008; Platzbecker et al. 2013; Pollyea et al. 2013; Ramsingh et al. 2013]. Furthermore, epigenetic therapy in the transplant setting seems to preferentially suppress GVHD while preserving GVL effects [Reddy et al. 2004; Choi et al. 2010], suggesting the potential for improving transplant outcome if used appropriately.
IMiDs and epigenetic drugs are not curative at this time, but leave an important treatment option for those who cannot tolerate conventional chemotherapy. It is natural to consider designing clinical trials combining one immunotherapy to the other, but the possibility of added toxicity should be monitored closely. IMiDs and epigenetic drugs have been shown to have effects on both side of immune regulation, activation and suppression, so it is important to find the dosage, timing and combination that allows their beneficial effects to prevail rather than immunosuppressive effects.
Strategies that block suppressor cells and hostile immune milieu against immune cells. Cyclophosphamide and 5-fluorouracil (5-FU) selectively suppress Tregs and MDSCs [Ghiringhelli et al. 2007; Vincent et al. 2010; Le and Jaffee, 2012], suggesting that these classical chemotherapeutic agents may have some effects via blockade of suppressor cell functions. As shown in the Figure 1, there are multiple immune evasion pathways that involve Tregs, for example, immune checkpoint engagement, and blockade of such pathways may be efficacious partly by indirectly abrogating the immunosuppressive capacity of Tregs. In murine AML models, Treg depletion by denileukin diftitox, a conjugated protein of IL-2 and diphtheria toxin, improved proliferation and the antileukemic effect of adoptively transferred cytotoxic T lymphocytes [Zhou et al. 2009] and denileukin diftitox prior to vaccination enhanced antileukemic T-cell response [Litzinger et al. 2007]. Treg depletion using daclizumab (an anti-CD25 antibody) has been demonstrated to improve vaccination potency in patients with solid malignancies [Jacobs et al. 2010; Sampson et al. 2012], but such data do not exist for AL. However, host Treg depletion by denileukin diftitox improved the efficacy of haploidentical NK cell infusions in AML patients, and translated into improved clinical outcome including better disease-free survival at 6 months [Bachanova et al. 2014].
MDSCs have also been investigated as a promising target for therapy. There are no agents that selectively deplete MDSCs, but numerous agents affect MDSC function [De Veirman et al. 2014]. For example, phosphodiesterase (PDE5) inhibitors augment antitumor response by MDSC suppression [Serafini et al. 2006]. An early clinical study of tadalafil in multiple myeloma suggested possible clinical benefit [Noonan et al. 2014] and there is an ongoing phase II trial testing tadalafil in conjunction with lenalidomide for the treatment of multiple myeloma [ClinicalTrials.gov identifier: NCT01858558]. Similar studies have not been performed for AL patients, but may be considered in conjunction with IMiDs or other immunotherapeutic modalities.
As discussed earlier, some of the immune evasion mechanisms mediated by IDO and arginine metabolites are intertwined with proliferation and function of MDSCs. In a preclinical model, a competitive inhibitor of IDO, 1-methyltryptopha (1-MT), and an oral small molecule that selectively inhibits IDO1, INCB024360, were shown to enhance antitumor response by T and NK cells, and prevent phenotypic conversion of T cells to Treg-like cells [Hou et al. 2007; Liu et al. 2010]. In the phase II study for patients with MDS, the agent was well tolerated but there was no significant changes in associated laboratory makers (such as intracellular IDO expression in mononuclear bone marrow cells and bone marrow MDSC percentage) or in clinical outcome [Komrokji et al. 2014].
The
Putting it all together – the role and timing of immunotherapy for AL
As a general principle, early phase studies with experimental agents can be justified only in the setting where there are no other better standard treatment options. As a result, immunotherapy for AL has been explored first in desperate settings such as post-transplant relapse and relapsed or refractory leukemia with high blasts burden. However, these are not the best situations in which immunotherapy can exert its role and the next step must be to define the best time to deliver immunotherapy. The timeline for intervention with immunotherapy in AL can be divided into four periods: prevention; remission induction; consolidation (maintenance of remission) including the option of HSCT; and the management of refractory or relapsed cases. Scheme of an example of how immunotherapy can be incorporated into the treatment course is depicted in Figure 3.

Timing of immunotherapy application in the course of leukemia progression.
Prevention
Unfortunately, although the increased risk of AML after prolonged immunosuppression in organ transplant recipients is evidence of immune control in preventing occurrence of leukemia [Adami et al. 2003; Vajdic et al. 2006; Villeneuve et al. 2007; Gale and Opelz, 2012], primary prevention of AL is unlikely to become the target of immunotherapy development considering the low disease incidence rate.
Immunotherapy at presentation and remission induction
At least in the near future, novel immunotherapeutic strategies will not be routinely incorporated as a part of induction therapy for newly diagnosed AL patients who can tolerate standard-of-care cytoreductive chemotherapy. In the setting of high leukemic blasts burden and a perturbed immune system, therapy exploiting the immune function is likely inadequate to control leukemia. However, this may not always be the case: CD19-CAR T cells directly target blasts and can eliminate high disease-burden B-ALL. The combination of immunomodulatory agents and intensive chemotherapy has been tested in limited numbers of studies for relapsed or refractory AL, and even less so in treatment-naïve AL. According to these limited data, concurrent usage of either lenalidomide or thalidomide with induction chemotherapy regimen does not improve the outcome but potentially increases toxicity [Cortes et al. 2003; Barr et al. 2007; Dennis et al. 2013]. The HDAC inhibitor, vorinostat, in conjunction with induction chemotherapy, has been shown to be tolerated and is active even in high-risk AML in phase I and II trials [Kadia et al. 2010; Garcia-Manero et al. 2012], although a long-term clinical impact of the combination remains to be demonstrated. Further incorporating cell-based therapy and other immune interventions in induction will require establishment of safety, efficacy and the pipeline to allow the cell products to be readily available for immediate clinical usage.
Immunotherapy for AL in remission or low disease burden
This may be advantageous in conjunction with consolidation chemotherapy or as a maintenance therapy after allogeneic HSCT to prevent relapse. For the control of any MRD that may be undetectable with current laboratory techniques, immune checkpoint inhibitors, IMiDs or hypomethylating agents could be helpful by boosting immune surveillance mechanisms. Numerous ongoing clinical trials are testing the concept of maintenance immunotherapy, augmenting immune surveillance during remission; anti-PD-1 [ClinicalTrials.gov identifier: NCT02275533], anti-PD-1 in combination with DC-AML vaccine [ClinicalTrials.gov identifier: NCT01096602], lenalidomide [ClinicalTrials.gov identifier: NCT01578954, NCT 02126553] and azacitidine [ClinicalTrials.gov identifier: NCT01757535, NCT01700673]. The bispecific antibody, blinatumomab, for B-ALL with persistent or recurrent MRD achieves durable complete remissions [Topp et al. 2011; Topp et al. 2012]. Cell-based therapies may also become a consolidation modality in the future.
Bridge to transplant
Various immunotherapies can be used as a bridge to transplant or as a part of the transplant preparative regimen. Most clinical experiences of cell-based therapy so far has been in refractory and/or relapsed cases without an HSCT option. In cases with HSCT options, cell-based therapy may achieve remission or at least MRD (as opposed to overt hematologic relapse) prior to transplantation, thereby improving the transplant outcome. Some cell-based therapies logistically require HSCT as a platform: CAR T-cell therapy for myeloid leukemia leads to marrow ablation as discussed above. It could only be used with transplant backup, unless reliable ways to control the in vivo persistence of adoptively transferred cells can be developed.
Post-transplant prevention of relapse
With improvement in transplant-related mortality over the past decades, the prevention of post-transplant relapse becomes important. The early post-transplant setting is uniquely advantageous for immunotherapy to work, where the leukemic burden is small and anti-leukemia lymphocytes have the chance to expand in the post-transplant immune milieu and lymphodepleted environment [Muranski et al. 2006]. Furthermore, it is possible to adoptively transfer donor cells that are not exhausted or tolerized as a part of transplant course. As described in the vaccine therapy section, post-transplant vaccines in conjunction with vaccine-primed lymphocytes to promote GVL are under investigation. Azacitidine for the treatment of MRD post-HSCT has been reported to either prevent or delay hematological relapse [Sockel et al. 2011; Platzbecker et al. 2012]. Prophylactic usage of low-dose azacitidine for patients with high-risk MDS/AML post-transplant is safe and may improve event-free survival and overall survival [de Lima et al. 2010]. IMiDs and immune checkpoint inhibitors are also being tested as a maintenance therapy post-transplant [ClinicalTrials.gov identifier: NCT01433965, NCT01254578] and CTLA-4 post-HSCT relapse [ClinicalTrials.gov identifier: NCT01822509].
Relapsed and refractory AL
Treatment of refractory/relapsed leukemia, especially post-transplant relapse, is challenging. This is the setting for experimental phase I immunotherapy studies and many examples of such trials are alluded to in this review. Breakthroughs achieved in immunotherapy treatment of such patients, notably with CAR cells, nevertheless support offering the opportunity for experimental treatments to even very poor prognosis patients.
Footnotes
Acknowledgements
AJB and KI are supported by the Intramural Research Program of the NHLBI, NIH.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Conflict of interest statement
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.