
Abstract
Background
Despite the great progress made in the last 10 years, alternative strategies might help improving definitive treatment options against hepatitis C virus infection.
Methods
With the aim of identifying novel inhibitors of the hepatitis C virus-1b replication targeting the viral NS3 helicase, the structures of previously reported symmetrical inhibitors of this enzyme were rationally modified, and according to docking-based studies, four novel scaffolds were selected for synthesis and evaluation in the hepatitis C virus-1b subgenomic replicon assay.
Results
Among the newly designed compounds, one new structural family was found to inhibit the hepatitis C virus-1b replication in the micromolar range. This scaffold was chosen for further exploration and different novel analogues were synthesised and evaluated.
Conclusions
Different new inhibitors of the hepatitis C virus genotype 1b replication were identified. Some of the new compounds show mild inhibition of the NS3 helicase enzyme.
Keywords
Introduction
Hepatitis C virus (HCV) is a primary cause of chronic liver disease and it affects 3% of the global population. 1 The infection, which becomes chronic in 60–85% of cases, leads to hepatic steatosis, fibrosis, cirrhosis and hepatocellular carcinoma.2,3 A vaccine is currently not available, while the standard of care was for long a combination of pegylated interferon and ribavirin. 4 HCV genome encodes six non-structural proteins, essential for the viral replication. 5 In the past 10 years, safe and highly potent inhibitors of the HCV replication have been developed, providing the basis for a definitive cure against this infection; their main targets are the NS3 protease,6,7 the NS5B polymerase, 8 the NS4B protein 9 and the NS5A protein. 10 Nevertheless, the available combination treatments with these agents are associated with high costs,11,12 and for each of them resistant viral strains have been reported.13–17
Despite the abundance of structural information on the HCV NS3 helicase, 18 none of the few inhibitors of this enzyme reported so far has been taken into clinical development. 19 This protein, essential for the viral replication,20,21 is responsible for the ATP-dependent unwinding of double-stranded RNA sequences, an intermediate in the viral nucleic acid synthesis.22,23
The NS3 helicase was chosen as target for the rational design and synthesis of novel potential agents against genotype 1b HCV. The structures of previously reported symmetrical inhibitors of the HCV NS3 helicase24,25 were rationally modified into four novel scaffolds, which were then synthesised and evaluated in the genotype 1b subgenomic replicon assay.
Results and discussion
Rational design and synthesis
The HCV NS3 helicase is formed by three domains and occupies the C-terminal portion of the NS3 protein. It presents multiple ligand-binding regions, the main ones being an ATP-binding site in the cleft separating domain 1 from domain 2 and a single-stranded nucleic acid-binding site at the interface of the three domains,
26
as depicted in Figure 1.
HCV NS3 helicase domains and main nucleic acid-binding cleft in the 3KQH crystal structure.
27
HCV: hepatitis C virus.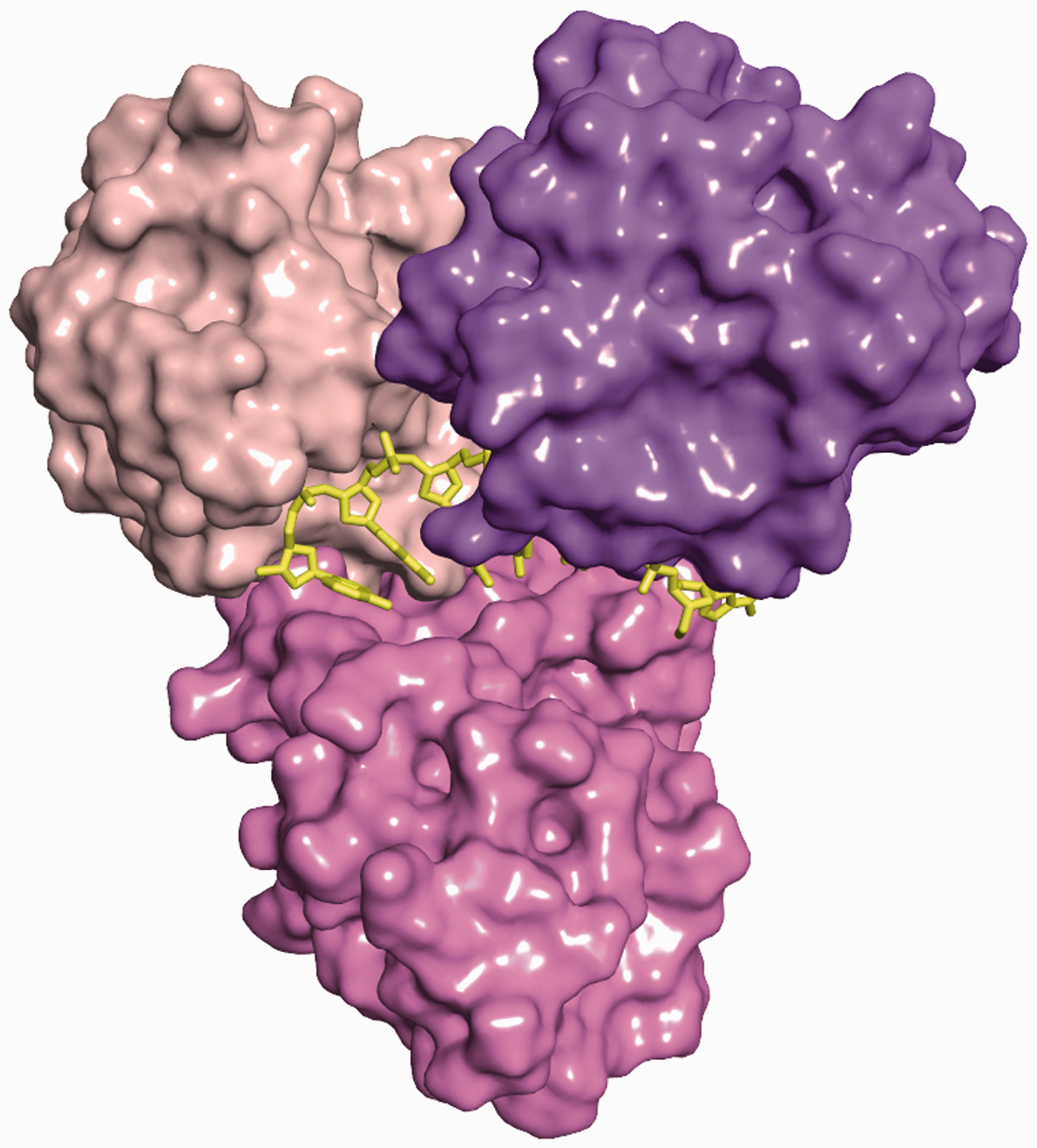
The potential interference with the known nucleic acid-binding cleft was evaluated for the design of novel inhibitors of the HCV replication. To achieve this result, the structures of previously reported symmetrical inhibitors of the HCV NS3 helicase were chosen as a starting point for the design of novel potential anti-HCV agents.24,25 Due to their bulky occupational volumes, the most likely site of interference with the enzyme is, for all these known compounds, the main RNA-binding pocket. In particular, as shown in Figure 1, the nucleic acid substrate occupies this pocket in an extended conformation in the high-affinity open structure of the enzyme, making interactions with residues belonging to all three helicase domains. Given their potential to interfere with the binding of the nucleic acid substrate of the enzyme, the structures of the known helicase inhibitors were rationally modified in order to combine their central linkers with two equal aromatic sulphonamide lateral groups. This last feature has been identified as important for the antiviral activity of a different series of agents against genotype 1b HCV recently found in our research group (unpublished data). The newly designed potential anti-HCV-1b scaffolds are described in Figure 2, along with the structures of the parent symmetrical helicase inhibitors.
Structure of the known symmetrical HCV NS3 helicase inhibitors considered and their proposed modifications. HCV: hepatitis C virus.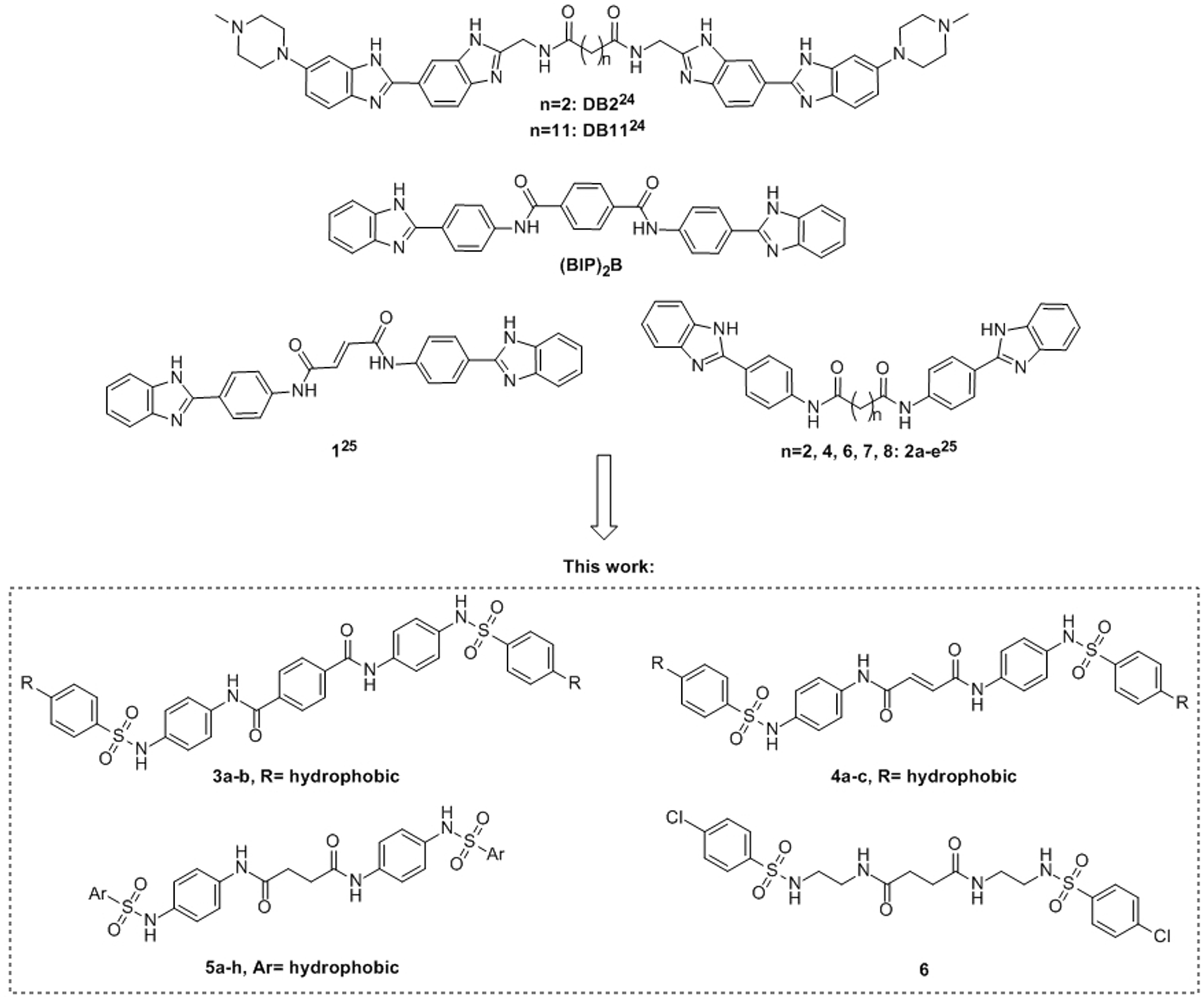
All four new scaffolds were analysed with molecular docking simulations with the Glide SP module in Maestro.
28
A 12 Å docking grid was generated from the 3KQH crystal structure, using as centroid the area defined by residues Arg393, Glu493 and Trp501. The presence of the newly inserted hydrophobic sulphonamides should maximise the potential interactions with the target site, as indicated by docking results (Figure 3(a) to (d)).
Predicted binding mode in the 3KQH crystal structure for the newly designed compounds: (a) 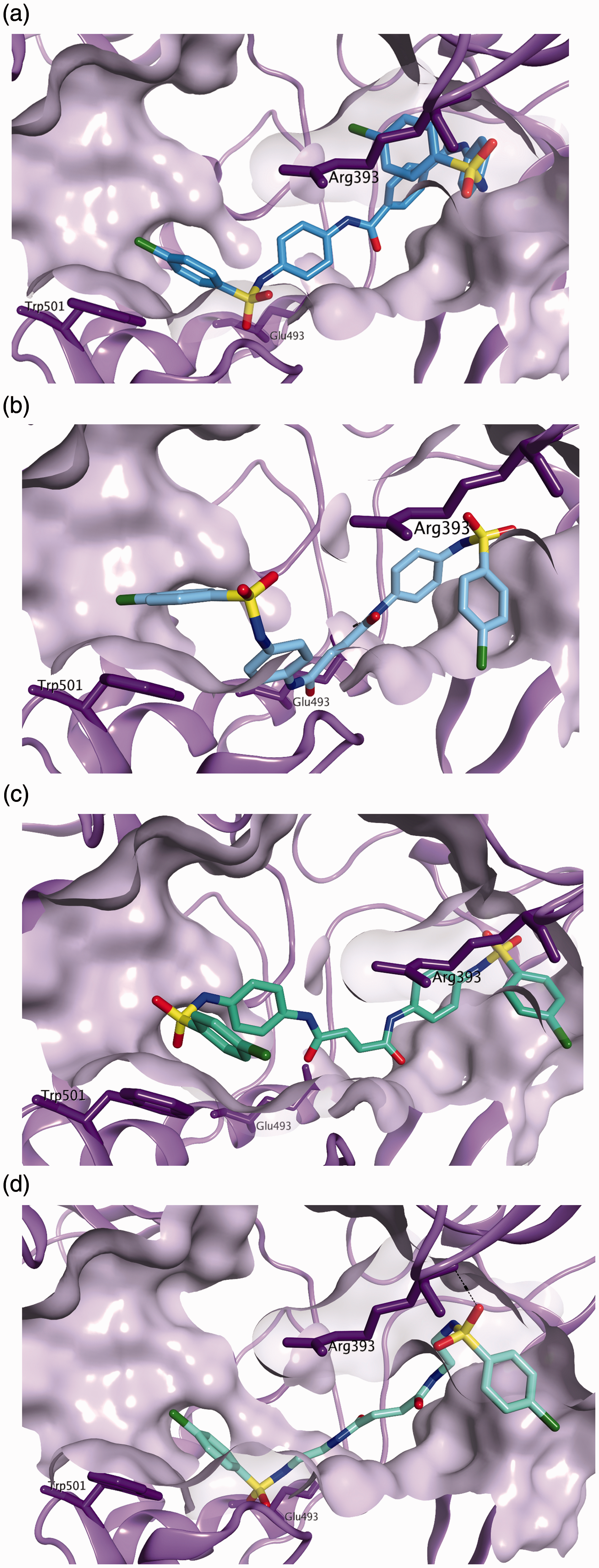
The predicted binding mode found for all the structures suggests a good spatial occupation of the target site of the HCV NS3 helicase, with an optimal fitting of the region at the interface of the three main domains, and with the potential of forming hydrophobic and hydrogen bond interactions with several residues, including Trp501, Arg393, Glu493, Thr411, Ser287, Asn556 and Phe557.
As docking results support the potential interference with the target site of the enzyme, all four new scaffolds were selected for synthetic development.
The new symmetrical structures were synthesised according to two different procedures, a general one for scaffolds
The common synthetic strategy applied for the scaffolds of Reagents and conditions: (i) DIPEA, anhydrous DCM, 0℃, 30 min, 53–93%; (ii) NEt3, anhydrous DCM, rt, 30 min, 36–69%; (iii) NEt3, anhydrous DCM, 0℃ 1 h, rt 2 h, 59%; (iv) NEt3, anhydrous DCM, rt 1 h, 78%. DCM: Dichloromethane; DIPEA: Diisopropylethylamine; NEt3: Triethylamine; rt: room temperature.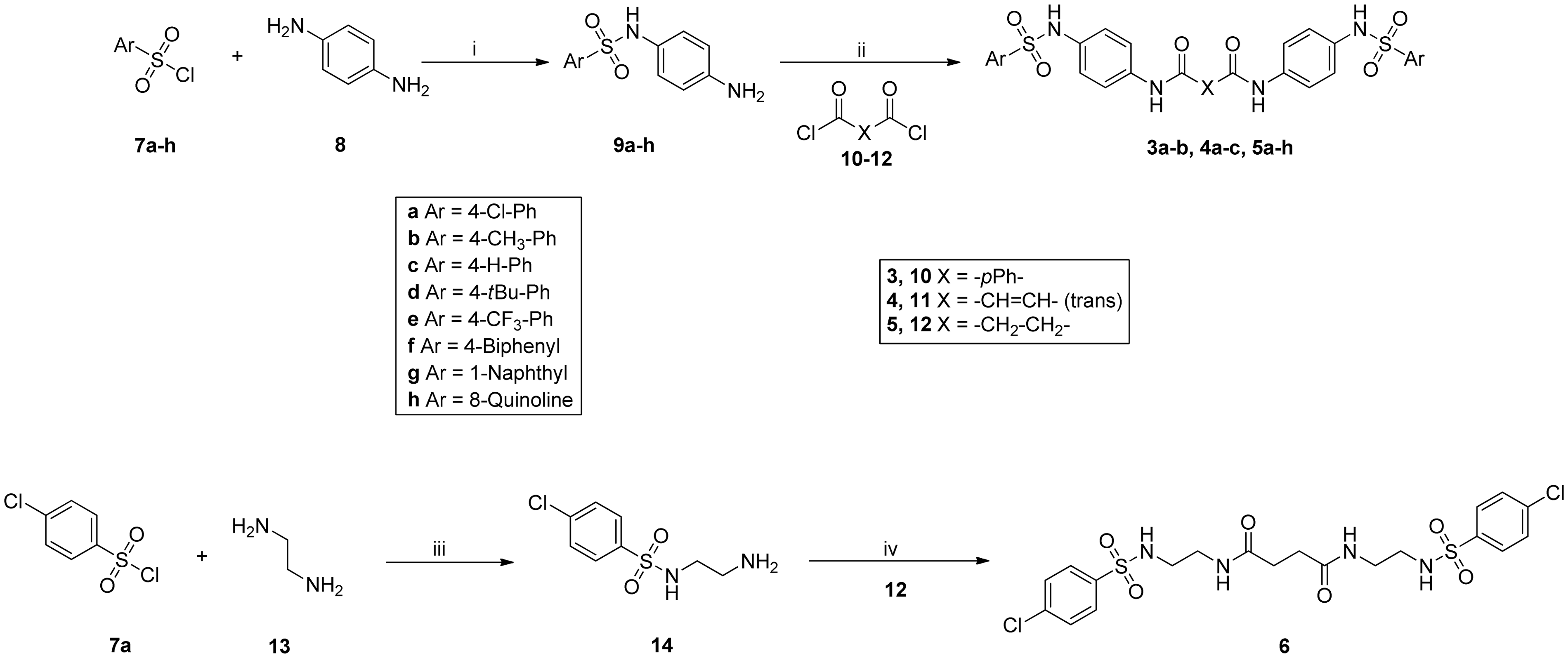
Common intermediates
Derivative
Biological activity
Antiviral effect of the test compounds on genotype 1b hepatitis C virus replication in the Huh5-2 replicon system.
EC50 = 50% effective concentration (concentration at which 50% inhibition of virus replication is observed).
The EC50, EC90, CC50 and IC50 values are the mean ± SD of at least 2–3 independent experiments.
EC90 = 90% effective concentration (concentration at which 50% inhibition of virus replication is observed).
CC50 = 50% cytostatic/cytotoxic concentration (concentration at which 50% adverse effect is observed on the host cell).
SI = the ratio of CC50 to EC50.
Among the four new structural families designed, the most interesting compound was initially found to be
Compounds
Conclusions
Starting from a rational approach, four new structural scaffolds were designed as potential inhibitors of the genotype 1b-HCV replication targeting the viral NS3 helicase. The newly designed compounds show the presence of the two equal phenylsulphonamide lateral groups, linked together with different symmetrical amide functions. For each new structural family, a small series of new derivatives was synthesised and evaluated in the HCV-1b subgenomic replicon and the helicase unwinding assay. One of these scaffolds showed antiviral activity in the micromolar range and was chosen for further development. Data found so far suggest that antiviral activity for these structures is associated with the presence of two equal phenylenediamine rings in the linker, connected by a central succinamide group. A hydrophobic substituent in the para position of the two terminal phenyl groups is important for activity retention. The best new succinamide analogues found,
Preliminary enzymatic evaluations suggest that the antiviral effect associated with the newly prepared compounds is mainly unrelated to the interference with the NS3 helicase activity, with other viral or cellular targets likely involved.
Experimental
All experimental procedures are described in detail in the Supporting Information, along with compound characterisation.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.