
Abstract
Teratocarcinosarcoma is a rare, highly aggressive malignancy of the head and neck, characterized by multiphenotypic and triphasic growth of epithelial, mesenchymal, and primitive neuroepithelial elements. Owing to its rarity and morphological heterogeneity, as well as the lack of experience with this neoplasm, teratocarcinosarcoma is often misdiagnosed, particularly in small biopsy samples when only some of the elements are identified, thus leading to delayed management. Aggressive clinical behavior and poor survival outcomes, necessitate an accurate diagnosis and appropriate treatment. This review describes the main demographic and clinicopathological features of teratocarcinosarcoma, with an emphasis on the recent advances that have attempted to identify the molecular signature of this neoplasm.
Introduction
Teratocarcinosarcomas (TCSs) are rare, highly aggressive, and peculiar malignant neoplasms of the head and neck that are notorious for their remarkable histological and biological heterogeneity. 1 The first case was reported in 1966 by Patchefsky et al., who described an ethmoid sinus tumor as a malignant teratoma. 2 This was followed by a report of three cases of paranasal sinuses tumors by Shanmugaratnam et al., who used the term “teratoid carcinosarcoma” to describe them in 1983. 3 One year later, Heffner and Hyams coined the term “teratocarcinosarcoma” to outline the complex pathological pattern that this neoplasm displayed in their case study. 4 In 2005, TCSs were recognized as a distinct entity by the World Health Organization Classification of Tumors: Pathology and Genetics of Head and Neck Tumors and were included in the “Carcinomas” section within the context of “Respiratory epithelial lesions” in the “Nasal, paranasal, and skull base tumors” chapter. 5 The overall incidence of TCS is extremely low, with fewer than 150 cases reported in the literature, most of which are single case reports or case series.6,7 It represents approximately 3% of all malignancies of the head and neck region and less than 1% of all cancers. 8 Familiarity with this entity is generally limited, and the exact classification of this tumor poses a real diagnostic challenge in surgical pathology, necessitating sufficient knowledge of its phenotypic diversity and specific diagnostic criteria. In this review, the epidemiology, clinicopathological features, treatment, and prognosis of TCS are discussed, with an emphasis on the recent developments that shed light on the molecular alterations and pathogenesis of this entity. This may serve as a useful reference for physicians and surgical pathologists unfamiliar with this tumor. Thus, a thorough literature review was carried out by searching PubMed, MEDLINE, and Google Scholar databases for all relevant English-language full texts and abstracts using the search queries “teratocarcinosarcoma” and “head and neck.”
Epidemiology and clinical manifestations
Teratocarcinosarcoma tends to affect adults with a mean age of 50 years; however, it has been reported in patients with a wide age range (1 month to 85 years).7–9 It shows a striking male predominance (approximately 83% of patients) with a male-to-female ratio of 7:1.6–8 The most common site of involvement is the superior aspect of the nasal cavity (approximately 79% of cases) with a predominance of the left side, followed by, in decreasing order of frequency, the paranasal sinuses, orbit, nasopharynx, pharyngeal wall, skull base, cribriform plate, and anterior cranial fossa.1,6,7,9–11 Nevertheless, rapid growth and extensive local destruction with direct extension into adjacent structures are frequently encountered at the time of diagnosis, and the site of origin cannot be determined in some cases.1,6 Although TCS shows a predilection for the sinonasal tract, in rare cases, tumors showing features resemble TCS have been reported to arise primarily in the thyroid gland and oral cavity.12–14
The clinical presentation depends essentially on tumor localization and the results of the mass effect, and usually has a short duration (average duration 3.5 months). The most common presentations are nasal obstruction, and epistaxis. Other presenting symptoms include headache, facial swelling, proptosis, blurred or loss of vision, eye and/or facial pain, epiphora, hypo/anosmia, expectoration of tissue, a mass protruding from the nostrils, and focal neurological deficits secondary to intracranial tumor extension.1,6,7,15 Infrequently, patients may present with signs and symptoms of the “syndrome of inappropriate antidiuretic hormone secretion.”1,16 Nasal endoscopic examination often reveals a variably gray-tan fleshy to reddish-brown hemorrhagic, friable, solid tumor mass with areas of necrosis. 6 TCS usually lacks specific radiological features and appears as a non-distinctive, large (mean size, 7.4 cm), locally destructive soft tissue mass of variable intensity.6,7,17
Pathological features
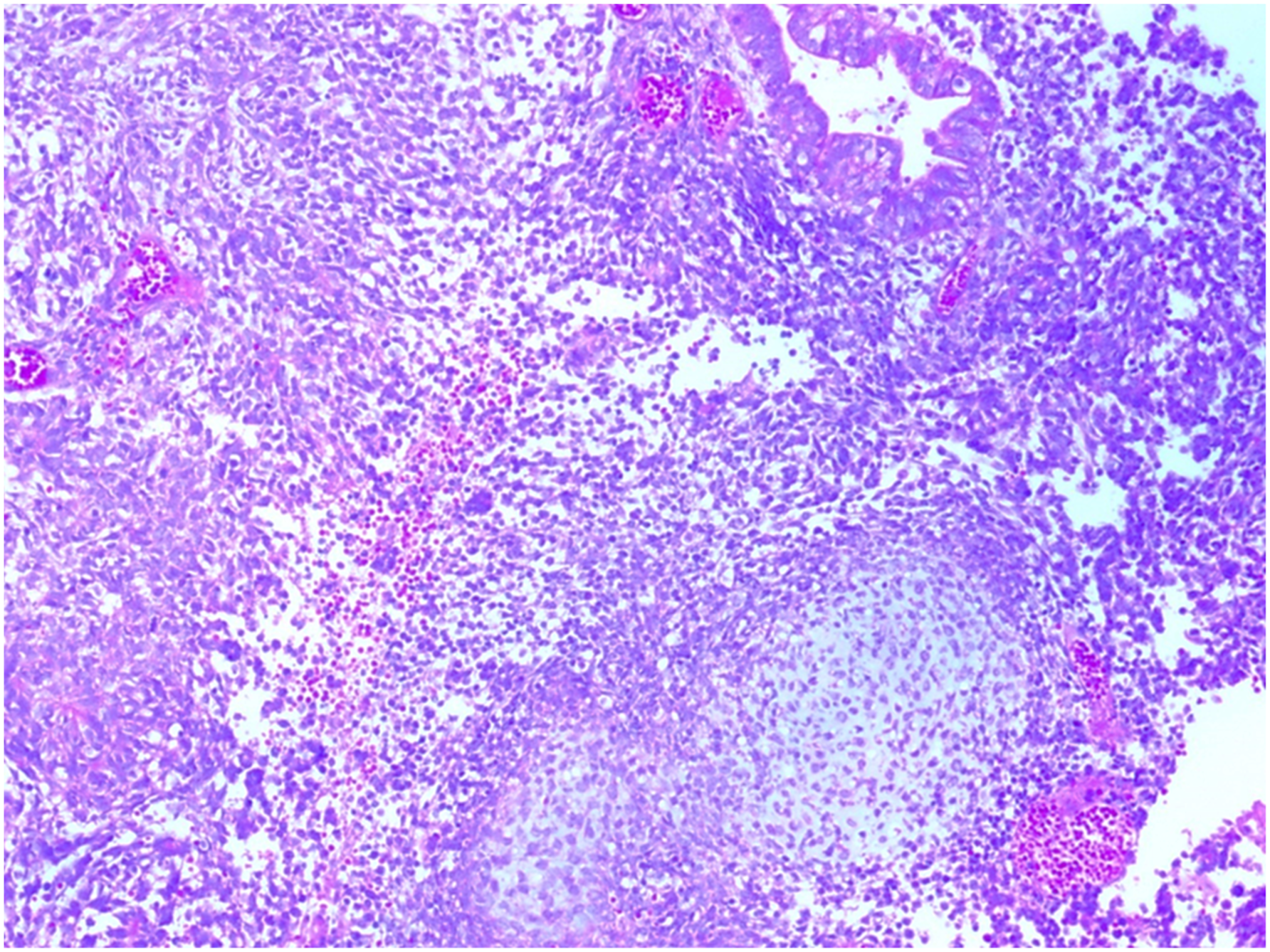
Teratocarcinosarcoma is characterized by a triphasic morphology comprising variable proportions of epithelial, mesenchymal, and neuroepithelial elements, showing varying degrees of maturation and cytologic atypia (Figure 1(a)). The epithelial component is highly variable and may be either squamous or glandular, featuring benign, malignant, mature and/or immature primitive morphologies. The squamous epithelium is mainly represented by nests and islands of cytologically bland, immature “fetal-appearing” clear squamous cells, which is a pathognomonic feature of this tumor. However, this finding is not consistent, as in rare cases the squamous epithelium may have a more mature appearance with prominent keratinization (Figure 1(b)).1,4,7,17,18 Glandular structures lined by benign-appearing and/or malignant intestinal-type mucinous or ciliated columnar respiratory-type epithelium are frequently seen, but to a quite variable extent (Figure 2).7,17,19 The mesenchymal-stromal component is also diverse and may be cytologically bland or overtly sarcomatous in appearance. This component can range from an undifferentiated, loose, and variably chondroid/myxoid-appearing stroma containing bland fibroblastic spindle cells with occasional periglandular accentuation, to a more cellular myoid-appearing mesenchymal tissue showing smooth muscle or rhabdomyoblastic differentiation with strap cells and cross-striations. Foci of osteoblastic, chondroblastic, and even high-grade or undifferentiated sarcomatous stromal elements are less common (Figure 3).1,7,11,17,19–21 The stromal and epithelial components are almost always intermingled with undifferentiated, typically high-grade, primitive neuroepithelial/neuroectodermal tissue comprising small blue cells exhibiting inconspicuous nucleoli and scant eosinophilic fibrillary cytoplasm, arranged in nests and islands, with variable foci of rosette structures and/or neuropil-like matrix (Figure 4).4,17,20,22 Occasionally, the neuroepithelial component morphologically resembles SMARCA4-deficient large-cell neuroendocrine carcinoma, featuring nested architecture with palisading of tumor cells, oval-to-carrot-shaped nuclei, a stippled chromatin pattern, and prominent nucleoli.
19
Furthermore, areas showing olfactory neuroblastoma-like morphology with uniform cytology, scant cytoplasm, round-to-oval nuclei, stippled chromatin, neurofibrillary matrix, and rosettes have been observed in some cases.
19
This neuroepithelial component may undergo chemotherapy-induced maturation with the formation of nodules of large ganglion-like cells embedded in an abundant neurofibrillary matrix, reminiscent of what may be encountered in a neuroblastomas or olfactory neuroblastoma.
23
Although germ cell components are consistently absent in TCS, foci resembling yolk sac tumors with papillary structures and hyaline cytoplasmic globules have been described.1,4,15,24 Likewise, TCS has never been described as a component of a mixed germ cell tumors.7,25 These observations emphasize that TCS is a unique multiphenotypic tumor of somatic origin, distinct from adnexal teratomas and genuine mixed germ cell neoplasms.25,26 As with other aggressive neoplasms, prominent mitotic activity and areas of tumor necrosis and apoptosis are frequently observed.
27
(a) Teratocarcinosarcoma exhibits admixture of primitive small round cells (thin arrow), squamous epithelium with clear cytoplasm (thick arrow), and glandular structures (asterisks) (hematoxylin-eosin, original magnification ×40). (b) At high power, the squamous epithelium shows cytologically bland fetal-appearing clear cells (left side) next to immature glands (right side) (hematoxylin-eosin, original magnification ×100). Neoplastic glands (upper side) intermingled with few smooth muscle fibers (arrows) that are embedded in a loose myxoid stroma (asterisks) (hematoxylin-eosin, original magnification ×100). A neoplastic gland is seen next to primitive neuroepithelial tissue (upper right corner) and chondroblastic-appearing stromal nodule (lower side) (hematoxylin-eosin, original magnification ×100). An island of fetal-appearing squamous epithelium with clear cells (thin arrows) is seen adjacent to a focus of primitive neuroepithelial tissue with a true rosette (thick arrow) (hematoxylin-eosin, original magnification ×100).


Immunohistochemically, TCS generally has a nonspecific immunophenotype that reflects the line of differentiation of various tumor elements, and the utility of immunohistochemical analysis is mainly for the identification of subtle components within the tumor.17,25 This includes detecting the expression of CKAE1/AE3, EMA, and CAM5.2 in the epithelial elements; p40 and CK5/6 within the squamous areas; CK7 limited to glandular structures; synaptophysin, chromogranin, CD56, neuron-specific enolase, CD99, GFAP, and INSM-1, highlighting the neuroepithelial component; desmin in myoid cells; and vimentin and p53 in a subset of cases.6,11,18,19,25,27,28 Skeletal muscle markers can aid the detection of subtle foci of rhabdomyoblastic cells. 21
Attempts have been made to identify sensitive and specific markers that reliably distinguish TCS from other sinonasal malignancies. Recently, immunohistochemical analysis of TCSs has revealed frequent loss of SMARCA4 (BRG1) protein expression in a majority (73% to 82%) of cases, with complete loss of nuclear staining across all tumor components in 60% to 68%, and partial/heterogenous loss in 14% of cases.19,26,29,30 SMARCA4 is a component of the switch/sucrose non-fermentable (SWI/SNF) chromatin remodeling complex that regulates the vital processes of cell proliferation and differentiation. 26 This marker may provide a useful and reliable diagnostic tool to confirm the challenging diagnosis of TCS in limited biopsy samples, particularly considering that SMARCA4 is intact in all other poorly differentiated sinonasal carcinomas and neuroendocrine tumors of the head and neck.26,29 Additionally, this raises the possibility that TCS may be part of the diagnostic spectrum with the novel entity “SMARCA4-deficient sinonasal carcinoma” and supports the hypothesis of origin from a multipotential stem cell with the capability of undergoing divergent differentiation.19,29,31
Another promising marker is SALL-4, a zinc finger transcription factor and common marker of germ cell tumors. It has been found to be relatively sensitive (85.7%) and specific (89.5%) for the diagnosis of TCS. However, its positivity is limited to the epithelial and primitive neuroepithelial elements and not to the mesenchymal or stromal components. 25 Although SALL-4 is expressed in sinonasal undifferentiated carcinoma and poorly differentiated neuroendocrine carcinoma to a much lesser extent than TCS, this promising marker appears to have great utility in distinguishing TCS from other high-grade, SALL-4 negative sinonasal tumors, including olfactory neuroblastoma and nasopharyngeal carcinoma. 25 Additionally, because SALL-4 shows a mechanistic association with the SWI/SNF complex, it may act as a surrogate immunohistochemical marker for the identification of alterations in complex proteins, including SMARCA4 loss. 25 In contrast to SALL-4 immunoexpression, β-catenin is overexpressed with significant nuclear localization, predominantly in the mesenchymal component but only in a subset of TCSs.19,25,30,32 Although approximately 83% of TCSs are positive for NKX2.2, its utility is limited when differentiating TCS from sinonasal tumors showing small round blue cell morphology, including Ewing sarcoma, melanoma, olfactory neuroblastoma, and small cell carcinoma. However, it may be useful to distinguish TCS from other sinonasal malignancies that do not express NKX2.2, including sinonasal undifferentiated carcinoma, SMARCB1-deficient carcinoma, and NUT carcinomas. 33 Immunoexpression of SMARCB1, SMARCA2, INI1, and Rb proteins is essentially intact, although most tumors show partial loss of SMARCA2, and a few cases show low levels or focal expression of the SMARCB1 protein.17,19,29
Ultrastructural studies of TCSs are limited. They have identified features resembling olfactory neuroblastomas, including the presence of neural processes with parallel microtubules in primitive stromal cells. Additionally, stromal cells show various degrees of skeletal muscle differentiation and a fibroblastic appearance, particularly in myxoid stromal areas. Tumors with squamous elements exhibit features characteristic of squamous differentiation, including desmosome-like junctions and intracytoplasmic tonofilaments.34,35
This complex and heterogeneous cytoarchitecture of TCS has led to considerable morphological and/or immunophenotypic overlap with entities under the spectrum of poorly or undifferentiated neoplasms of the sinonasal tract, which poses a diagnostic challenge, particularly on limited biopsy material because not all components may be present.25,26 Thus, an adequate and representative biopsy specimen is essential for accurate diagnosis, which requires the recognition of all tumor components.
SMARCA4-deficient neoplasms, such as SMARCA4-deficient sinonasal undifferentiated carcinoma and SMARCA4-deficient sinonasal neuroendocrine carcinoma, are important differential diagnoses because they share a common molecular pathogenesis and frequent neuroendocrine marker expression (though usually focal and weak), despite morphological heterogeneity and they are associated with poor overall survival.19,26 TCS is often mistaken both radiologically and pathologically, particularly with limited biopsy samples, with other malignant neoplasms of the head and neck, including poorly differentiated squamous cell carcinoma, adenocarcinoma, malignant craniopharyngioma, olfactory neuroblastoma, small cell carcinoma, sinonasal undifferentiated carcinoma, rhabdomyosarcoma, chondrosarcoma, malignant salivary gland tumors, and carcinosarcoma/sarcomatoid carcinoma. The latter is characterized by a biphasic appearance with a variable combination of malignant epithelial and sarcomatous elements, in contrast to the triphasic appearance of TCS.4,6,18,36 On the other hand, metastatic melanoma of the head and neck rarely shows TCS-like dedifferentiation with complete loss of melanocytic markers. Recognition of this phenomenon is important to keep in mind since the management plan for malignant melanoma is totally different from the management plan for TCS. 37 Moreover, SMARCA4-deficinet malignancies in different organs that occasionally metastasize to the head and neck, such as SMARCA4-deficient undifferentiated lung carcinoma, should be considered in the differential diagnosis of TCS. 26 The precise distinction between TCS and these neoplasms has significant prognostic and therapeutic implications.
Pathogenesis and molecular features
The pathogenesis of TCS has remained controversial for decades; however, a possible origin from pluripotent progenitor or stem cells in the olfactory or sinonasal mucosa, capable of divergent differentiation, has been postulated.1,4,7,18,29,38,39 However, an origin from primitive embryonic tissues that remain sequestered in the sinonasal tract has also been proposed.3,4 Furthermore, the defining genetic abnormalities of this mysterious tumor received significant attention over the last few years. Recent genetic studies have made major advances in this regard through the identification of frequent biallelic somatic inactivating mutations of SMARCA4, a tumor suppressor gene, and a member of the SWI/SNF chromatin remodeling family of proteins. This alteration has been observed in 65% of studied cases and is considered the dominant genetic event in TCSs. In 29% of cases, this genetic alteration is often associated with a simultaneous CTNNB1 activating point mutation.9,19,29–32 Another, but less common, genetic abnormality in TCS is a somatic CTNNB1 (β-catenin) gene activating point mutation (p.S45F codon), which has been reported in 35% of studied cases.30,32 This provides useful insights into the potential genetic driver mutations of TCS, particularly when considering that both the SWI/SNF complex and Wnt/β-catenin pathway are implicated in the tumorigenesis of this neoplasm. Other less frequent genetic alterations that may have a role in TCS molecular pathways involve other members of the SWI/SNF family and the Wnt/β-catenin pathway. These include inactivating SMARCB1 mutations in 6%, simultaneous SMARCB1, APC, and ARID1A mutations in approximately 6%, and DICER1 hot spot mutations in 17% of studied TCSs. 30 Amplification of chromosome 12p13, although absent in evaluated TCS cases, has been described in a subpopulation of tumor cells in a TCS case, but the significance of this remains unknown.15,24 In TCS case, a hyperdiploid clone characterized by trisomy 12, with an additional subclone showing del (1p), has been identified through conventional cytogenetic analysis. 27 Another case of TCS identified through multigene panel sequencing somatic mutations in PIK3CA gene. 40 The significance of these findings and the roles of chromosome 12 alterations and PIK3CA mutations in the molecular pathogenesis of TCS remain largely unknown and require further investigations.
Treatment and prognosis
Historically, TCS has shown highly aggressive biological behavior. The outcome and overall survival are generally poor, with an overall survival of 46% and a death rate of 29%.6,7,39 Most TCS cases show an average disease-free survival rate at 2 years of 28% to 55%.8,39 Unfortunately, local recurrence is frequent (22% to 38% of patients, with a mean time to recurrence of 19.5 months) and the metastatic rate is high (8% to 11%), with 8% of patients developing both local recurrence and distant metastasis.6–8 The most frequent distant metastatic sites include the lungs, dura, cervical lymph nodes, and intracerebral metastasis.8,11,41–43
Due to the aggressive nature of this neoplasm, multimodal therapeutic approaches should be considered. Although there are no official guidelines, the most common treatment strategy currently used for TCS is a bimodal therapy consisting of radical surgical resection with adjuvant radiotherapy, which is the strategy selected in 52% to 62% of cases.6,7,42 The second most common management option is a trimodal approach combining surgery with adjuvant chemotherapy and focused radiation therapy, which has been used in 28% of cases. 7 This trimodal strategy appears to be the optimal choice, as it has shown the highest survival and lowest recurrence rates, as well as a significantly delayed time to death compared with bimodality or single surgical options.7,8,39 Of the individual treatment modalities, radical surgical resection is the most common choice and has been used in 87.2% to 90% of patients, followed by adjuvant radiation therapy or chemotherapy in 59.3% of patients.3,6–8 Less common treatment options include surgery with adjuvant chemotherapy in 20% of patients, surgery without adjuvant treatment, radio-chemotherapy, neoadjuvant chemotherapy with surgical resection, and surgery with intensity-modulated radiation therapy. 6 There are no standard surgical guidelines; however, the reported surgical techniques include maxillectomy, lateral rhinotomy, ethmoidectomy, sphenoidectomy, and open craniofacial resection, which can be performed using an open, endoscopic, or a combination approach.1,8,32 However, intracranial tumor seeding may complicate surgical resection, in addition to the difficulty in achieving complete tumor resection and adequate margins in most of cases.32,44 Postoperative intensity-modulated radiation therapy may achieve optimal dose distribution and improve the outcomes of patients with TCS.8,45 Alternatively, proton beam therapy can provide satisfactory results in some patients. 46 Neoadjuvant chemotherapy with a “cisplatin and etoposide protocol” may be considered in patients with unresectable TCSs. 44 In patients with p.S45F β-catenin mutations, targeted inhibitors of β-catenin may be applied following adequate future trials of these medications. 32 Given the dismal course of TCS, new therapeutic paradigms must be identified.
Conclusions
Although TCS is a site-specific sinonasal malignancy, occurrence at other sites of the head and neck would merit replacing the current name “sinonasal teratocarcinosarcoma” with a broader one, such as “teratocarcinosarcoma of the head and neck.” Historically, TCS has been a management challenge for physicians owing to its rarity, aggressive behavior, and lack of standardized therapeutic protocols and guidelines. Larger studies are required to reach a consensus on the appropriate management algorithms. Comprehensive mutational analysis and sequencing of a large number of cases are required to gain further insight into the genetic pathways governing TCS. The identification of the underlying genetic drivers may provide potentially successful adjuvant or targeted medical therapy options for this lethal neoplasm.
Footnotes
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.o