
Abstract
We present a case of a 61-year-old woman with relapse/refractory multiple myeloma post-autologous peripheral blood stem cell transplantation who presented with recurrent febrile episodes and infection. Worsening thrombocytopenia during lenalidomide-based therapy for early biochemical relapse warranted further investigations. Even though it was initially attributed to lenalidomide and relapse/refractory multiple myeloma, there was not much improvement in her counts despite adjustment of her medications. Subsequent serial bone marrow results and cytogenetic evolution eventually revealed therapy-related myelodysplastic syndrome progressing to therapy-related acute myeloid leukemia. In view of her age and comorbidities, she was treated with subcutaneous azacytidine with palliative intent which resulted in transient disappearance of blasts in the peripheral blood. She subsequently succumbed to progressive therapy-related acute myeloid leukemia and relapse/refractory multiple myeloma. This case highlights the complexity in diagnosing therapy-related myelodysplastic syndrome earlier as prompt diagnosis is crucial in delaying its progression to therapy-related acute myeloid leukemia. Managing therapy-related acute myeloid leukemia in the setting of relapse/refractory multiple myeloma post-autologous peripheral blood stem cell transplantation is challenging in terms of balancing the treatment toxicity and providing the best possible quality of life.
Introduction
The treatment landscape for multiple myeloma (MM) has evolved and improved significantly over the years. While this translates to better outcomes and survival, it also carries the risk of therapy-related myeloid neoplasm (t-MN), which includes therapy-related myelodysplastic syndrome (t-MDS), therapy-related acute myeloid leukemia (t-AML) and t-MDS/myeloproliferative neoplasm. 1 t-AML is defined as the diagnosis of acute myeloid leukemia with a history of previous cytotoxic chemotherapy and/or radiation therapy. 1 Published data on the management and outcomes in t-MN developing in relapse/refractory multiple myeloma (RRMM) patients, particularly in the post-autologous peripheral blood stem cell transplantation (PBSCT) setting is limited. RRMM is defined as a disease which becomes non-responsive or progressive on therapy or within 60 days of the last treatment in patients who had achieved a minimal response or better on prior therapy. 2 Here we report our experience in managing a patient with RRMM post-autologous PBSCT who developed t-AML with an antecedent history of myelodysplastic syndrome (MDS). This case highlights the complexity of concomitant t-MDS progressing to t-AML and RRMM, when clinical management in balancing both conditions could be challenging. Early suspicion and detection with prompt diagnosis and management may halt the progression of this otherwise poor prognostic condition.
Case report
A 61-year-old woman with underlying childhood bronchial asthma, hypertension and dyslipidemia presented with a 3-week history of persistent right thigh pain in June 2012. She was diagnosed with immunoglobulin (Ig)Gκ multiple myeloma (Durie Salmon stage IIIA and International Staging System stage III). Her blood investigations showed hemoglobin of 9.4 g/dL, white blood cell (WBC) counts of 6.2 × 109/L, platelet counts of 285 × 109/L, corrected calcium of 2.53 mmol/L and serum creatinine of 86 µmol/L with estimated glomerular filtration rate (Modification of Diet in Renal Disease; MDRD) 63.6 mL/min/1.73 m². Peripheral blood film (PBF) showed mild rouleaux formation without abnormal plasma cells. Serum and urine protein electrophoresis (SUPE) showed the presence of a monoclonal band in the gamma region of 16.2 g/L and a faint monoclonal band of 13.4 mg/L, respectively. Bone marrow aspirates (BMA) revealed 16% abnormal plasma cells. Cytogenetic analysis for cultured bone marrow showed normal karyotype (46,XX). Fluorescence in-situ hybridisation (FISH) analysis using Vysis LSI IGH dual color break apart rearrangement probe, Vysis TP53/centromere 17 (CEP17) FISH probe kit, spectrum orange probe Vysis LSI D13s319/13q34 FISH probe kit showed no abnormalities. The serum β2-microglobulin level was 6.2 µg/mL while the lactate dehydrogenase level was 390 U/L (normal range 125–220 U/L).
Initial treatment with cyclophosphamide, thalidomide, dexamethasone and zoledronic acid for six cycles resulted in very good partial response (VGPR) with serum paraprotein of 1.8 g/L and absence of urine paraprotein. She underwent peripheral blood stem cell mobilisation with intravenous cyclophosphamide 3 g/m2 in November 2012 with total harvested 15.5 × 106 CD34+ cells/kg. Autologous PBSCT was performed in January 2013 following conditioning with intravenous high-dose melphalan (200 mg/m2); CD34+ cell dose was 10.3 × 106/kg. She was in complete remission (CR) for the next 3 years but refused maintenance therapy. In October 2015, she developed biochemical relapse in which her serum paraprotein quantitation was 2.9 g/L with immunofixation electrophoresis showing IgGκ. As she was asymptomatic, she was monitored closely and her serum paraprotein was stable, ranging between 2.7 and 5.3 g/L (Figure 1).

Timeline of the clinical course, laboratory results (serum paraprotein, hemoglobin and platelet), treatment with serial bone marrow aspirates and cytogenetic results based on MM disease status. Timing of administration of medications is shown. CDTZ: cyclophosphamide, dexamethasone, thalidomide, zoledronic acid; Ld or Len/dex: lenalidomide/dexamethasone; MM: multiple myeloma; VTD: bortezomib/thalidomide/dexamethasone.
In October 2016, she developed intermittent right pelvic pain while serum and urine paraprotein increased to 16.5 g/L and 42.2 mg/L. She was treated with lenalidomide 25 mg on days 1 to 21 and oral dexamethasone 40 mg on days 1, 8, 15 and 22 (Ld) for four cycles. However, treatment was changed to bortezomib, thalidomide and dexamethasone (VTD) for three cycles as lenalidomide supply was interrupted. Once available, Ld was resumed for another 10 cycles. She subsequently achieved VGPR. Her SUPE was performed at another centre in between her follow-ups at our hospital as she had to travel from a different state during this period. She began to develop worsening thrombocytopenia, ranging between 30 and 50 × 109/L since February 2018 (Figure 1), which was presumably due to lenalidomide. Other differential diagnoses included t-MDS and immune thrombocytopenia. Despite reducing lenalidomide to 25 mg every other day and treatment with elthrombopag with a maximum dose of 100 mg daily, her platelet counts did not recover. BMA in July 2018 showed 6% abnormal plasma cells with dysplastic changes. Karyotyping revealed normal female chromosome, but FISH analysis showed the presence of three copies of TP53 and CEP17 in 43% of the cells analysed (Figure 2(a) and (c)). In view of the persistent thrombocytopenia and symptomatic relapse MM, treatment was changed to bortezomib, thalidomide and dexamethasone (VTD) regime. This was continued to January 2019 with her serum paraprotein ranging between 3.5 and 6 g/L and an absence of urine paraprotein.

Conventional cytogenetic analysis showing (a) normal female karyotype at diagnosis in 2012 and in July 2018; (b) deletion of the long arm of chromosome 7 (black arrow) from BMA sample in April 2019. (c) Fluorescence in situ hybridisation (FISH) analysis using Vysis TP53/CEP17 FISH probe kit from BMA sample showing three orange and three green signal pattern which indicated three copies of TP53 and CEP17 in July 2018 (43%) and April 2019 (15%). BMA: bone marrow aspirates.
In January 2019, although she was asymptomatic, her serum paraprotein increased to 10.1 g/L. Unfortunately, she decided to continue with the VTD regime. However, in April 2019, she presented with gluteal abscess and her blood investigations showed bicytopenia with haemoglobin of 8.5 g/dl, WBC of 13.5 × 109/L, absolute neutrophil counts of 12 × 109/L and platelet counts of 16 × 109/L. Unfortunately, PBF showed 4% blast cells. There were 14% abnormal plasma cells in BMA and increased blast cells (13%) with dysplastic changes in a hemodiluted sample. Trephine biopsy confirmed excess of blast cells with diffuse infiltration of plasma cells (Figure 3). Immunophenotyping analysis of BMA sample showed the presence of both myeloblasts and abnormal plasma cell populations. Cytogenetics analysis revealed an abnormal female chromosome complement with deletion of the long arm of chromosome 7 with breakpoints in 7q32 as the sole anomaly in all cells examined (Figure 2(b)). FISH analysis showed the persistent presence of three copies of TP53 and CEP17 in 15% of the cells analysed (Figure 2(c)). The IgGκ level increased to 17.1 g/L. Given the serial BMA changes and cytogenetics evolution, the most likely diagnosis at this stage was t-MDS transforming to t-AML with concomitant RRMM.
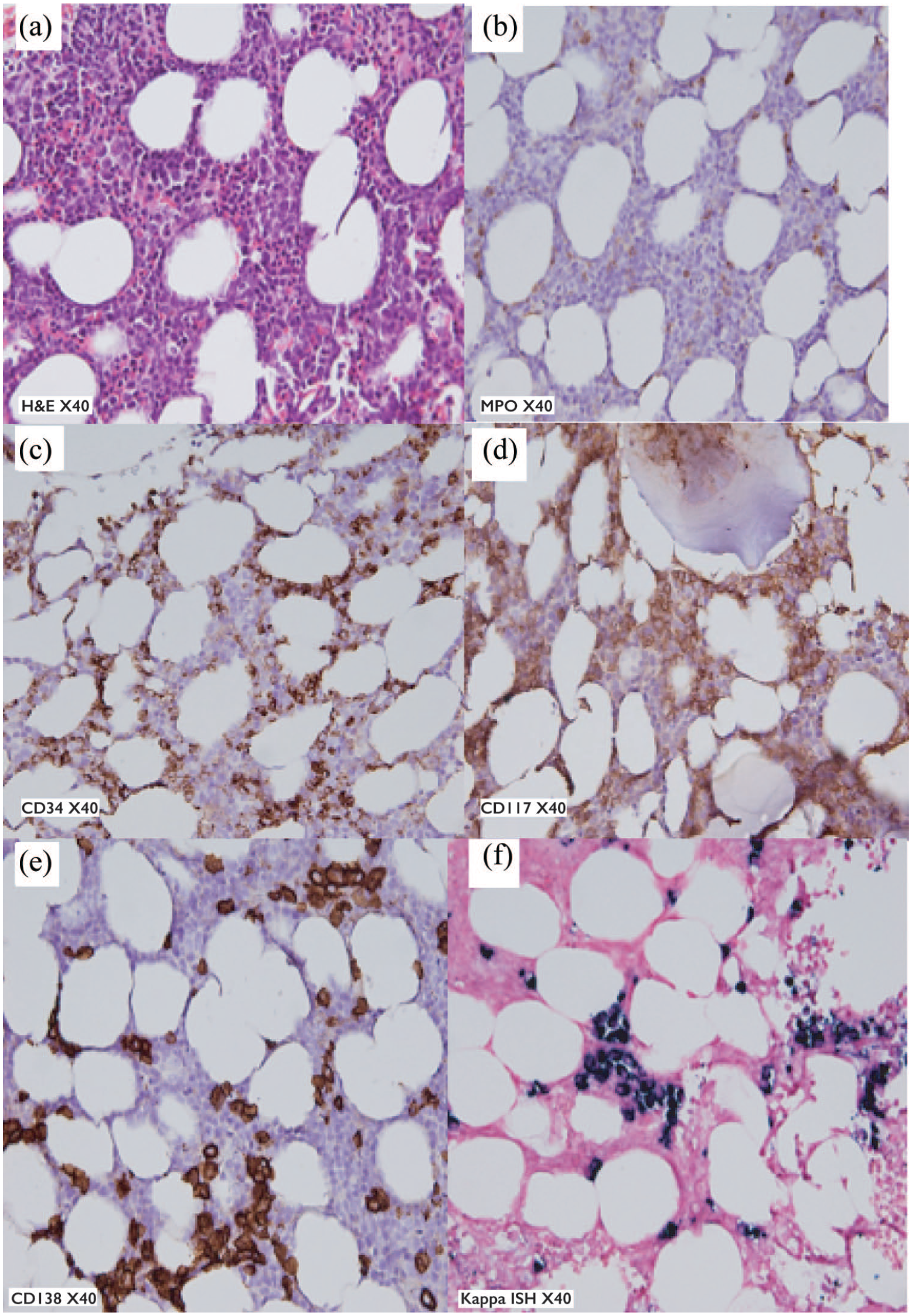
Photomicrograph of bone marrow trephine biopsy performed in April 2019 showing (a) clusters of medium size blasts cells as well as patchy infiltration by plasma cells which exhibited eccentric nuclei (haematoxylin and eosin staining, ×40, original magnification); blasts cells staining (b) negative with myeloperoxidase (×40, original magnification); (c) positive for CD34 (×40, original magnification); (d) positive for CD117 (×40, original magnification); (e) clusters and interstitially distributed plasma cells (CD138 immunohistochemical staining, ×40, original magnification); (f) the plasma cells express kappa light chain restriction confirming their clonality (kappa in-situ hybridisation, ×40, original magnification).
Our patient opted for treatment with palliative intent in view of her age and comorbidities. Treatment with subcutaneous azacytidine 75 mg/m2 for 7 days every 4 weeks was commenced. There was disappearance of blasts in the peripheral following two cycles of treatment while a repeat BMA showed 12% abnormal plasma cells and 12% blast cells. However, her disease substantially progressed while on the third cycle and she succumbed to her illness in October 2019.
Discussion
There is scarcity of published data on t-MN occurrence, clinical characteristics and outcomes in the Asian population, particularly with post-autologous PBSCT. The onset of cytopenias during re-commencement of Ld therapy at biochemical relapse warrants further investigations to ascertain the aetiology. Differential diagnoses included drug-induced, infection-related, relapse MM and t-MN. The presence of dysplastic changes in the BMA performed in July 2018 were not specific for t-MDS as it could also be due to idiopathic cytopenias of undetermined significance, idiopathic dysplasia of undetermined significance, clonal haematopoiesis of indeterminate potential and clonal cytopenias of undetermined significance, thus raising the dilemma in the diagnosis of t-MDS. Features that may suggest t-MDS against others include the severity of cytopenia and its relation to the timing of previous chemotherapy and/or radiotherapy, and most importantly cytogenetic changes. Thus, it is imperative to monitor peripheral blood films in the setting of cytopenia and performing cytogenetic analysis especially from the bone marrow sample as MDS-associated cytogenetic abnormalities (CA) may precede clinical disease by many months. MDS-CA is defined as cytogenetic abnormalities typifying MDS. 3
The incidence of t-MN in MM patients following chemotherapy was reported to be between 0.2% and 7%.4 –6 The possible risk factors for t-MDS in our patient included exposure to high-dose chemotherapy at a relatively younger age (54 years) during the diagnosis of MM, which was followed by autologous PBSCT, IgG subtype of MM and RRMM status. These patient and disease-related factors were further augmented with MM-related therapy, namely alkylating agents (cyclophosphamide and melphalan) and immunomodulatory agents (thalidomide and lenalidomide), which further contributed to clonal selection and damage to bone marrow microenvironment. Gertz et al. reported that 27% had active MM at the diagnosis of MDS/acute myeloid leukemia. 7 MM relapse was reported to be one of the risk factors for MDS-CA, 8 while few studies found that the majority of MM patients were in stringent CR, CR or at least partial remission.5,9 Few studies reported advanced age, longer duration of pretransplantation chemotherapy, low CD34 yield, and more leukapheresis sessions to harvest the desired quantity of haemopoietic progenitor cells are found to be predictive of t-MN.4,5,8,9 However, our patient had good yield, 10.3 × 106/kg CD34+ cell infusion in one leukapheresis session prior to autologous PBSCT. The duration from autologous PBSCT to the diagnosis of t-AML in our patient was 6 years, consistent with the reported time from MM diagnosis/treatment to the diagnosis of t-MN, which ranged from 4 to 12 years.10,11 Given this latency time, older patients may have died prior to the development of t-MN as they usually have worse MM-related survival.
Different chemotherapeutic agents exert their cytotoxic effects through different mechanisms and these further cause cytogenetic aberrations, genomic instability and damage. Our patient developed t-MDS with extra copies of TP53 and CEP17 via FISH 8 months prior to the diagnosis of t-AML. TP53 encodes for p53 tumour suppressor protein. TP53 has been reported in 20–50% of patients with t-AML and is associated with unfavourable cytogenetics, an increased number of chromosomal abnormalities, intrinsic therapy resistance and worse prognosis.12,13 Prior exposure to cytotoxic agents or radiation therapy leads to TP53 mutations which are associated with genetic instability, thus leading to deletion 5q and/or complex karyotype in patients with t-MN. 14 The development of del7q in this patient may have accelerated the progression to t-AML. Cases with deletion of the long arm of chromosome 7 (–7/del7q) typically occurred following treatment with alkylating agents and are associated with a latency period of 4–5 years, a myelodysplastic prodrome and chemotherapy resistance. 15 Usmani et al. observed MDS-CA in 11% of the cases in their cohort of patients and this often preceded clinical MDS/acute leukemia. 8 In a study involving 301 t-AML patients, Sasaki et al. reported five t-AML patients whose primary diagnosis was MM in which two had an antecedent history of dysplasia. 16 With the evidence of antecedent dysplasia, it would be interesting to determine whether these patients would benefit from earlier disease-modifying treatment for MDS to halt the progression to AML.
There is no consensus on the treatment of t-MDS and t-AML in the RRMM setting to date. Careful consideration based on the age at disease onset, comorbidities, MM status, tolerability to previous therapies, the predominant disease at current presentation, eligibility for allogeneic haematopoietic stem cell transplantation (HSCT) and patients’ preference are imperative before deciding on the definitive treatment in this poor prognosis condition. Several combinations which include conventional chemotherapy, hypomethylating agents (HMAs) and monoclonal antibodies have been described in previous papers, with survival ranging between 2 and 17 months.4,7,11 In the absence of clinical trials in a resource-limited country, low-intensity chemotherapy or best supportive care may be offered to patients unfit for aggressive therapy. Our patient was treated with azacytidine which resulted in transient stable disease and a considerably good quality of life. Gertz et al. reported no clear evidence of response in the seven patients on HMAs in their study. 7 In the setting of MDS/AML with TP53 mutation, Welch et al. reported that all 14 patients achieved various levels of CR with 10 days of decitabine therapy. 17
Conclusion
We report this case due to its rarity and complexity in getting the diagnosis of t-MDS which subsequently progressed to t-AML, the possible contributing factors for t-MN in a RRMM patient and therapeutic challenges. Development of a risk prediction model will be beneficial for clinicians to identify MM patients who are at risk of developing t-MN. The detection of these events at an earlier stage will further set the course of the treatment towards an improvement of progression-free survival in the future.
Footnotes
Acknowledgements
The author(s) would like to thank the Cell Therapy Center, Hospital Canselor Tuanku Muhriz and the Dean of the Faculty of Medicine, UKM for support in writing this manuscript.
This case had been presented as a poster presentation at the 17th Annual Scientific Meeting of the Malaysian Society of Haematology, 10–12 September 2020, Kuala Lumpur, Malaysia.
Authors’ contributions
DNS, FRJ and NRT collected data and drafted the manuscript; SAA and SS provided the photomicrographs with descriptions; SAA and SS helped review the manuscript and provided feedback for improvement. All authors have read and approved the manuscript.
Availability of data and materials
Data sharing is not applicable to this article, as no datasets were generated or analysed during the current study.
Conflict of interest
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical approval
Ethical approval was not sought for the present case report because it is not required as per university guidelines. This study was completed in accordance with the Declaration of Helsinki.
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Informed consent
Written informed consent was obtained from legally authorised representatives before the study.