
Abstract
Objective
Protein kinase C-delta (PKC-δ) is involved in apoptosis. This study aimed to establish whether PKC-δ can further promote IL-1β-induced chondrocyte apoptosis by mediating the phosphorylation of the JNK and p38 mitogen-activated protein kinase (MAPK) signaling pathways In osteoarthritis (OA).
Methods
We employed chondrocyte staining to determine the extent of cartilage degeneration. PKC-δ and p38 signal expressions were used in the immunohistochemical (IHC) test and apoptosis was assayed at the TUNEL test in human osteoarthritic and controls. We stimulated rat cartilage cells using IL-1β (10 ng/ml)/rottlerin (10 μM) or lentivirus. To determine the apoptosis rate, we employed flow cytometry. The mRNA of both BCL2-related X (BAX) and cysteine aspartate protease 3 (caspase-3) could be measured via qRT-PCR. Western blot measured the protein levels of BAX, caspase-3, PKC-δ, p-JNK/JNK and p-p38/p38.
Results
The positive rate of PKC-δ and the apoptotic rate of chondrocytes in OA were higher than controls. The manifestation of PKC-δ was positively related to the degree of cartilage degeneration, p38 protein expression, and apoptosis rate. IL-1β exposure upregulated PKC-δ expression in chondrocytes in a dose-dependent manner. Decreasing PKC-δ expression and its phosphorylation in OA can inhibit MAPK signaling pathway activation (phosphorylation) by downregulating JNK and p38 protein phosphorylation and expression. This inhibition decreases caspase-3 and BAX levels, consequently lowering the apoptosis rate in chondrocytes.
Conclusion
PKC-δ activation by IL-1β in OA promotes chondrocyte apoptosis via activation of the JNK and p38 MAPK signal pathways, thereby promoting the OA progression.
Introduction
In orthopedic clinics, osteoarthritis (OA) is as a frequent chronic refractory joint injury. The function and life quality of OA patients are seriously threatened due to the high rate of handicap. 1 Articular cartilage damage and degeneration are the main features of OA. The resident cells of joint cartilage consist only of chondrocytes. Chondrocyte apoptosis is an effective pathological change in OA. As OA is intimately linked to chondrocyte apoptosis, this process is expected to be a novel target for OA manipulation and alleviating cartilage damage and degeneration. 2 In response to a variety of stimuli, chondrocytes undergo phenotypic changes and express a series of factors, from which a vicious cycle begins to destroy cartilage and trigger an inflammatory process. 3 In addition, pro-inflammatory factors (PIC) are essential mediators in the inflammatory response; interleukins (IL), the tumor necrosis factor (TNF), as well as matrix metalloproteinases (MMP) also lead to the occurrence and development of OA.4,5 To date, however, the specific mechanisms of OA pathogenesis are not fully understood, and OA treatment is still slow and ineffective. As a result, it is critical to continue researching new treatment targets of OA to further explore more effectual healing strategies.
Protein kinase C (PKC) refers to a family of protein kinases that depend on phospholipids and calcium ions. PKC plays a role in a wide range of biological actions including cell growth, cell differentiation, programmed cell death, autophagy, and the metabolism of multiple cells, including chondrocytes, via its own signaling pathway or in conjunction with other signaling pathways.6,7 It can also modulate the synthesis and degradation of cartilage extracellular matrix and the metabolic loop of inorganic pyrophosphate in cartilage, being one of the key signal transduction molecules affecting cartilage growth, development, and degeneration.8,9 PKC-β1, PKC-α, and PKC-ζ are the PKC isoforms closely related to cartilage, along with PKC-δ. PKC-δ and PKC-ζ were found to be expressed in articular cartilage tissue of OA patients in tissue microarray data. 10 PKC-δ, one of the elements of the novel PKC network, can activate and regulate cell signal transduction pathways through specific phosphorylation sites, thereby achieving a variety of biological effects.11,12 IL-1, a classical pro-inflammatory factor, also participates in the enablement of PKC-δ, and the expression levels of PKC-δ have a dose-dependent effect on IL-1. 13 These findings suggest a correlation between the severity of inflammatory response and the expression and activation of PKC-δ in OA. Activated PKC-δ acts as an essential mediator of the apoptosis signal, mediating apoptosis through the pathways of mitogen-activated protein kinase (MAPK), NF-κB, p53, and ROS-activated transcription factor and the mitochondrial and nuclear translocation of PKC-δ. The expression and activation of PKC-δ and its relationship with apoptosis are more widely studied in tumors, such as prostate cancer, 12 non-small cell carcinoma, 14 chondrosarcoma, 15 and adult T-cell leukemia. 16 OA research remains minimal locally and internationally. Currently, the signal transduction pathways of chondrocyte apoptosis are multiple and sophisticated, among which the MAPK pathway is the main upstream signal transduction pathway causing chondrocyte apoptosis. 17 The MAPK pathway involves the MAP kinase (MPK), MAPK kinase (MEK, MKK or MAPK kinase), and MEK kinase (MEKK, MKKKK or MAPK kinase). Upon stimulation by growth factors, inflammation, hormones, as well as endogenous stress and environmental signals, MAPKs receive activation signals from MKKs and MKKKs, are activated in response, and manifest as cascading phosphorylation. 18 MAPKs are traditionally classified into 2 major categories: mitogenic and stress-activated MAPKs. The classical representatives of mitogenic responses are ERK MAPKs which have notable effects in tumor multiplication, transfer, and aggression; tumor extracellular matrix degradation; and tumor angiogenesis. In addition, JNK and p38 MAPKs are response elements to stressful stimuli, and the JNK & p38 MAPK signal pathways play vital roles in responses to stress, such as inflammation and apoptosis.19-21 The c-Jun amino-terminal kinases (JNK) consist of JNK1, JNK2, and JNK3. In neoplasms, JNK is affected by chemoresistance to positively regulate autophagy to counteract apoptosis; in joint diseases, however, JNK and its downstream cascades function in chondrocyte apoptosis by facilitating inflammation, with JNK as a pro-apoptotic signal under endoplasmic reticulum stress conditions.22-24 p38-α, p38-β, p38-γ, and p38-δ are the 4 isomers of p38 MAPK. The p38 MAPK pathway is not only a classical inflammatory pathway but is also one of the upstream pathways by which activated cysteine aspartate proteases (caspases) induce cell apoptosis. p38 affects the stability of BCL2 members by phosphorylation cascades and targeting related factor pathways at the transcriptional level, resulting in its pro- or anti-apoptotic function.25,26 BAX, a pro-apoptotic gene, is ubiquitous in the cytoplasm. With enhanced expression of BAX and activation of the mitochondrial pathway, cytochrome c is liberated and initiates apoptosis of chondrocytes in OA in a caspase-3/-7-dependent manner. BAX inhibition abrogates chondrocyte apoptosis due to lysosomal dysfunction, and OA chondrocyte apoptosis exhibits a robust correlation with BAX.27,28 Activated caspases are one of the molecular hallmarks of apoptosis. 29 Caspase-3 was overexpressed in cartilage from patients with OA during immunohistochemistry and TUNEL assays, demonstrating that caspase-3 is associated with the progression of OA. 30 Caspase-3 may cleave dead substrates and lead to apoptosis resulting in the destruction of chondrocytes, ultimately contributing to the progression of OA. 31 Both in chondrocytes from human OA patients and in IL-1β-induced OA in rat articular chondrocytes, the pro-apoptotic effect was found to be related to increased BAX protein levels and caspase-3 activation, suggesting that Bax and caspase-3 inhibition may be an approach to delaying articular cartilage degeneration.32,33 JNK and p38 MAPK signaling pathways regulate cellular processes comprising cell differentiation, proliferation, survival, apoptosis, and inflammation. 34 The JNK and p38 MAPK signal pathways have a dual function during apoptosis and act as activators or inhibitors according to the cell type, stimulus, environment, tissue, and organism involve.17,35 The specific roles of their pro- or anti-apoptotic mechanisms are complex, and more investigations are needed to resolve those conflicts. In OA, phosphorylation of the JNK and p38 MAPK signaling pathways and their downstream transcriptional progenitor sets activate and function in chondrocyte apoptosis, providing clues to the substrates that act upon PKC-δ expression and activation in OA. 36
In this study, the expression and apoptosis of PKC-δ and p38 proteins of the MAPK signaling pathway in human osteoarthritic cartilage tissues and normal articular cartilage tissues were investigated. We used the lentiviral knockdown of PKC-δ and PKC-δ inhibitor rottlerin to assess their effects on chondrocyte apoptosis. We discovered that IL-1β activation of PKC-δ in OA can promote chondrocyte apoptosis through the JNK and p38 MAPK signaling pathways and thus promote OA progression, a finding that offers a fresh idea and theory foundation for the clinical treatment of OA.
Materials and Methods
Antibodies and Reagents
Peprotech (Rocky Hill, NJ, USA) supplied us with recombinant rat IL-1β. PKC-δ inhibitor rottlerin was obtained from MedChemExpress (Monmouth Junction, NJ). Abcam (Cambridge, MA, USA) offers PKC-δ (ab182126), JNK (ab124956), and p38 (ab32142) antibodies. The Cell Signaling Technology (Beverly, MA, USA) provided antibodies against p-PKC-δ (2055S) and p-p38 (4511S). Proteintech (ProteinTech Group, Chicago, IL, USA) provided antibodies against p-JNK (80024-1-RR), BAX(50599-2-Ig), caspase-3(66470-2-IG), and GAPDH (10494-1-AP).
Collection and Assessment of Human Articular Cartilage Samples
All donors have provided verbal informed consent for the use of cartilage samples of human origin. The Ethics Committee of the First Affiliated Hospital of Guangxi Medical University has endorsed the study (2016-KY-114). Cartilage tissue from 30 OA cases (7 mild, 9 moderate, and 14 severe) and 11 normal articular cartilages were collected and stained hematoxylin–eosin (H&E), Saffron-O/fast green, and Masson cartilage stain (Solarbio, Beijing, China). Then, the degree of cartilage degeneration was quantified using a modified Mankin score. The expression of the key genes of the PKC-δ and MAPK signaling pathways and chondrocyte apoptosis in human osteoarthritic cartilage and normal articular cartilage were assessed by immunohistochemistry and TUNEL assay.
Rat Primary Chondrocyte Culture
Four 3- to 7-day-old SD rats were killed by cervical dislocation. After death, the rats were immersed in 75% alcohol for 5 minutes, and then their articular cartilages were dissected under aseptic conditions. The articular cartilages were collected and minced by microscopy, digested with trypsin-EDTA digest (0.25%) (Solarbio, Beijing, China) at 37°C, and then instilled with collagenase type II at a concentration of 0.2% for 4–6 hours. The digested cartilage was resuspended and inoculated with Dulbecco’s Minimum Essential Medium (which contains 10% fetal bovine serum and 1% penicillin mix) in Petri dishes. The cells were allowed to grow in a humid environment at 37°C with 5% CO2. Thereafter, the substrate was replaced every 2 to 3 days. The cells were observed to increase to around 80% density when digested using a 0.25% trypsin-EDTA digest, then were passaged in culture at a particular density. The morphology of P0 to P2 cells did not change significantly. Therefore, second-generation chondrocytes were used in all in vitro experiments.
Chondrocyte Intervention
To investigate the expression levels of PKC-δ signaling, the chondrocytes were subjected to IL-1β (0, 2.5, 5, 10, and 20 ng/ml) for 48 hours at differing concentrations, and the optimal action concentration was selected. In an in vitro functional study of PKC-δ, chondrocytes were pretreated with IL-1β (10 ng/ml) for 48 hours to simulate an inflammatory environment and then infected with lentiviral LV-shRNA-NC and LV-shRNA-PKC-δ for 24 hours; by contrast, chondrocytes were treated with rottlerin (10 μM), an inhibitor of PKC-δ, for 24 hours.
Lentiviral Transfection
GeneChem (Shanghai, China) supplied us with the lentiviral LV-shRNA-PKC-δ and LV-shRNA-NC. The lentiviral transfection of chondrocytes was added at a cell confluence with 30%–50%. After 24 hours, the medium was changed when the survival rate was > 95%. The cells were observed to be > 90% transfected under fluorescence microscopy after 3 days. The cells were passaged and cultured for subsequent experiments, and the effect of transfection was measured by qRT-PCR and Western blot.
Immunohistochemistry (IHC), TUNEL Test, and Histological Staining
Cartilage tissue specimens were immobilized in 4% paraformaldehyde for over 48 hours, followed by decalcification via 10% ethylenediaminetetraacetic acid in paraffin wax for 4 to 6 weeks. Individual tissue samples were sliced into 4 μm sections and then individually stained with H&E, Saffron-O/fast green, and Masson cartilage cell staining solution. The extent of cartilage degeneration was quantified by 2 observers using a modified Mankin score. After paraffin sections had been dewaxed, EDTA high pressure antigen repair, 3% hydrogen peroxide incubated at 37 °C for 10 min to eliminate endogenous peroxidase activity, followed by soaking overnight at 4°C with primary antibody (PKC-δ diluted 1:200, p38 diluted 1:200). Primary antibody substituted with PBS as a negative control. The following day, incubation was performed by immersion with secondary antibodies and concluded with DAB staining and hematoxylin staining. The sections were dehydrated, fixed with neutral adhesive, visualized, and recorded under a microscope. All image results were interpreted according to staining intensity and percentage of positivity. TUNEL Apoptosis Detection Reagent Box (Roche) was used to detect the apoptosis level of bone tissue.
Western Blot Assessment
Total chondrocyte protein was isolated by mixing high efficiency RIPA lysate (tissue/cell) (Solarbio, Beijing, China), PMSF (Solarbio, Beijing, China), phosphatase inhibitor (ComWin Biotech Co., Ltd. Beijing, China), and protease inhibitor (MedChemExpress, Monmouth Junction, NJ) to form a protein lysis buffer. The protein lysis buffer was added to the cells and then placed on ice for at least 30 minutes before centrifuged for 30 minutes at 4°C and 12,000×g. Protein density was determined with the Omnieasy™ Instant BCA Protein Assay Kit (Shanghai, China). The proteins were separated by electrophoresis, transferred, encapsulated in nitrocellulose filter membranes (Merck KGaA, Darmstadt, Germany), and closed with protein-free fast blocking buffer (Shanghai, China) for 30 minutes. The membranes were soaked and hatched in primary antibody overnight at 4°C. The next day, the membranes were incubated with secondary antibodies under room temperature for 1 hour. Washings were performed thrice at 10 minutes each using Tris-Buffered Saline with Tween 20. The proteins were visualized using chemiluminescence reagents (Invitrogen, Carlsbad, CA, USA) and the Wes system from ProteinSimple. The gray scale values of the immunoblots were measured for quantitative analysis with the Image 3.0 software (BioRad).
Quantitative Real-Time Reverse Transcription Polymerase Chain Reaction (qRT-PCR)
RNA was obtained in rat chondrocytes with the RNAeasy™ Animal RNA Isolation Kit (Beyotime Biotechnology, Shanghai, China). Reversal transcription of 1,000 ng RNA was achieved with the PrimeScript™ RT Master Mix (Takara, Beijing, China). Then, qRT-PCR assay was undertaken with cDNA as a template. The Biosystems™ Power SYBR™ Green (ABI, Los Angeles, CA, USA) probe was used, together with a specific primer sequence synthesized by Biotech Engineering Services (Shanghai, China). For the endogenous control, GAPDH was selected to normalize the PKC-δ exposure, and the comparative exposure of PKC-δ was computed by the 2−ΔΔ Ct method. The primer pair of the target gene is shown in Table 1 .
Primer Sequences Used in This Study.
Flow Cytometry
Cell apoptosis was assessed with the Annexin V-APC/7-AAD apoptosis reagent box (Kogan Biotechnology, Jiangsu, China). Initially, 2 x 106 cells were placed in centrifugal tubes and washed with pre-cooled PBS as specified. The cells were then resuspended by adding 500 μl of 1 x binding buffer. Following 30 minutes incubation on cold ice, 5 μl/tube of annexin V-APC and 10 μl/tube of 7-AAD were added to the suspended cells for approximately 5 minutes under light-proof conditions. The last flow cytometry (Becton, New Jersey, USA) was performed, and the results were assessed via FlowJo_V10.
Analysis of Statistics
All analyses were completed using SPSS software program 25.0 (IBM, Armonk, NY, US). The data were presented as mean ± standard deviation, with independent samples t test for 2 group comparisons and 1-way ANOVA for multigroup comparisons. The expressions of PKC-δ and P38 proteins in OA tissue and normal cartilage tissue were analyzed with the chi-square test or the Fisher precise probability method. Spearman’s rank correlation analysis was applied to evaluate the relationship between the (1) PKC-δ protein expression levels and the degree of OA cartilage degeneration and (2) p38 protein expression levels and correlation with TUNEL staining scores. Data were regarded as statistically significant if P < 0.05.
Results
PKC-δ and P38 Proteins are Hyper-Expressed in Human Osteoarthritic Cartilage Tissue
Articular cartilage specimens from normal and OA patients were taken for histological analysis. Immunohistochemical analysis revealed significantly higher positive rates for PKC-δ and p38 proteins in the MAPK pathway in human osteoarthritic cartilage than in normal articular cartilage (
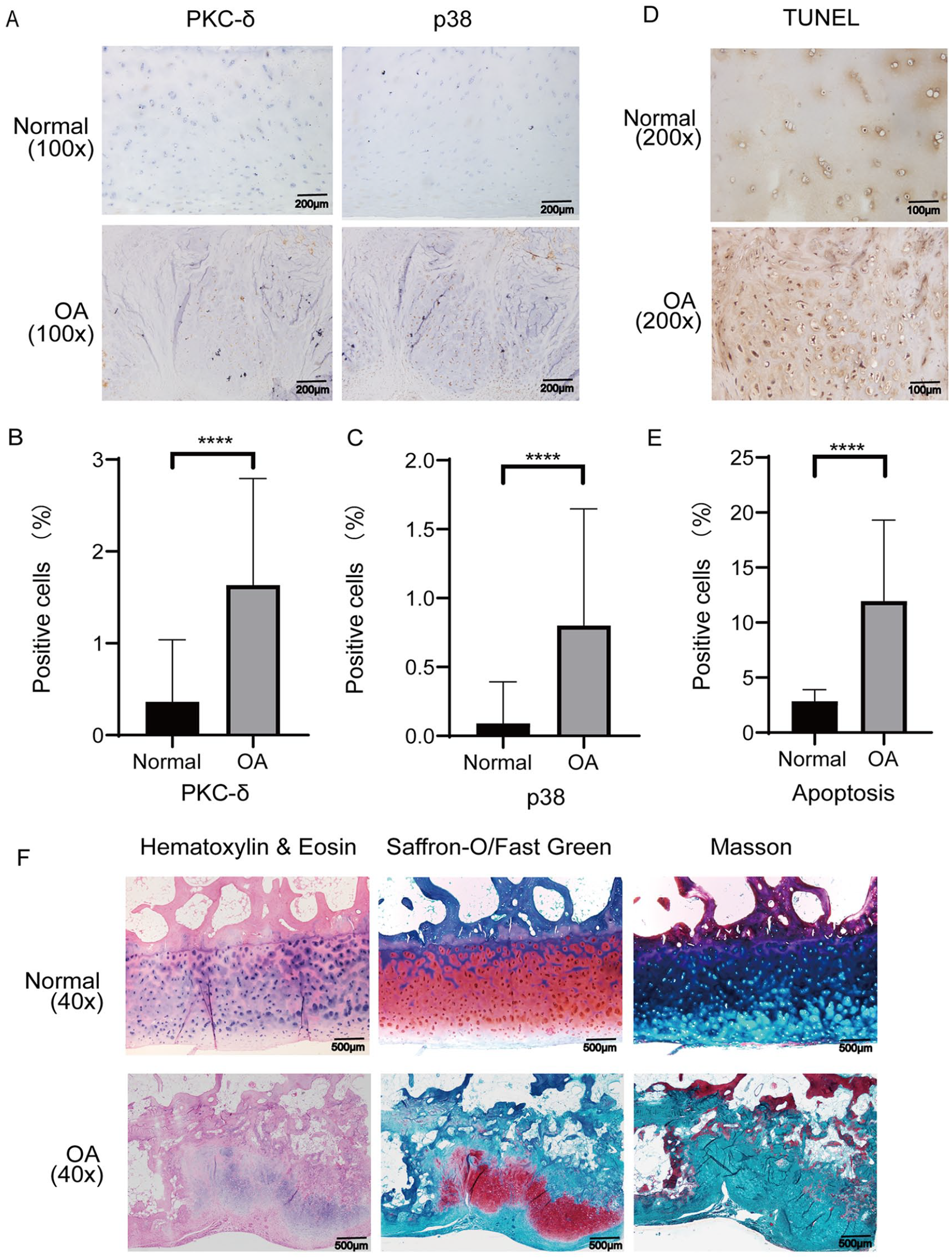
Staining evaluations of PKC-δ and P38 in human OA cartilage and normal articular cartilage and staining of chondrocyte apoptosis. (
Saffron-O/fast green, H&E, and Masson dyeing showed degeneration of articular cartilage in OA (
Effects of IL-1β and PKC-δ Inhibitor Rottlerin on Chondrocytes
The CCK8 detection indicated that different concentrations of IL-1β (0, 0.625, 1.25, 2.5, 5, 10, 20, 40, 80, and 160 ng/ml) were not cytotoxic to SD rat chondrocytes (
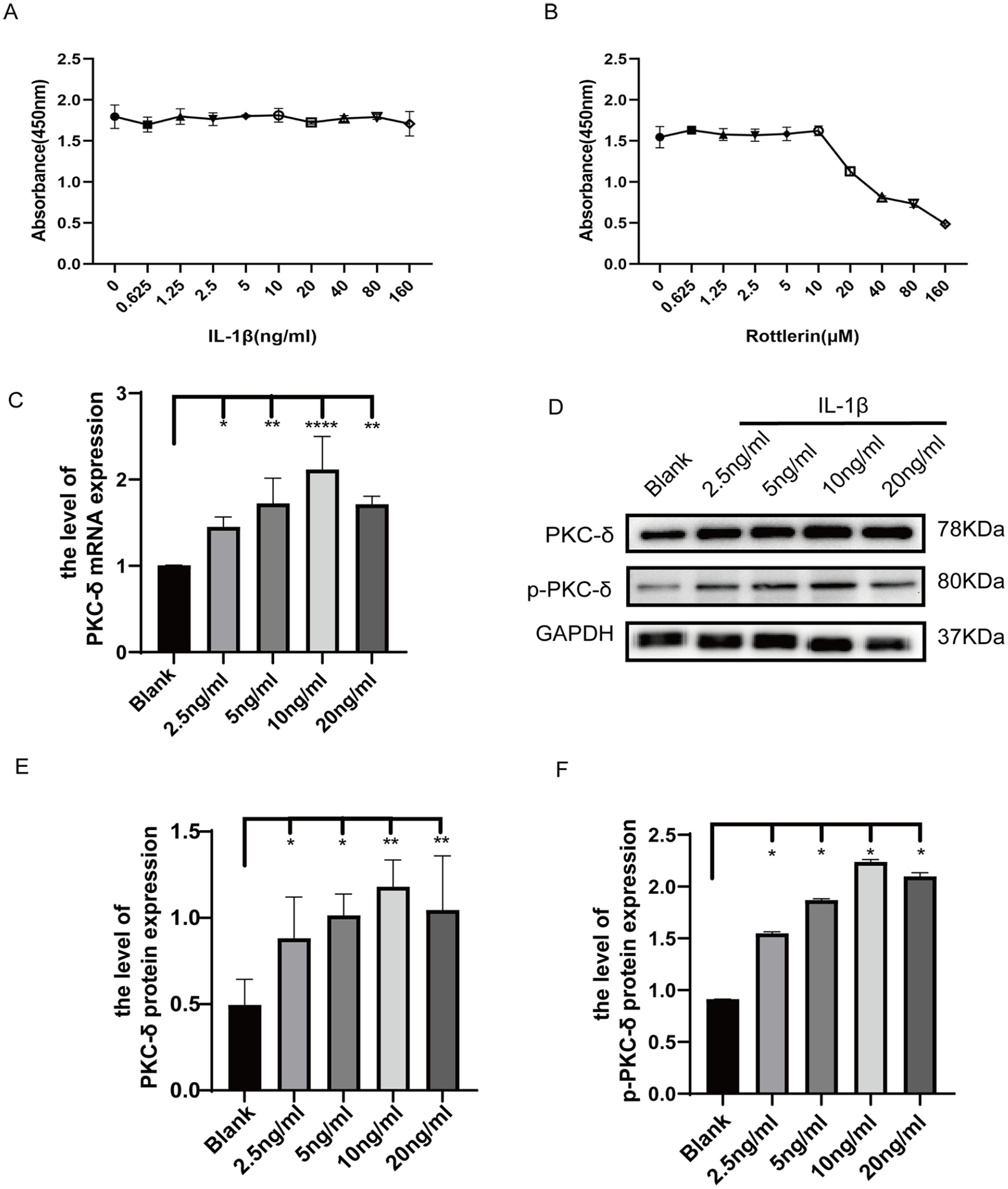
PKC-δ and p-PKC-δ expressions are increased in IL-1β-triggered chondrocytes. (
Effect of IL-1β-Induced Inflammatory Response on PKC-δ
qRT-PCR assessment showed that IL-1β exposure upregulated PKC-δ expression in chondrocytes in a dose-dependent pattern (
Involvement of PKC-δ in Chondrocyte Apoptosis in OA
To probe the influence of PKC-δ on apoptosis in OA chondrocytes, we used lentivirus (LV-shRNA-PKC-δ) to silence PKC-δ in chondrocytes and subsequently observed green fluorescence expression in more than 90% of the cells under inverted fluorescence microscopy (
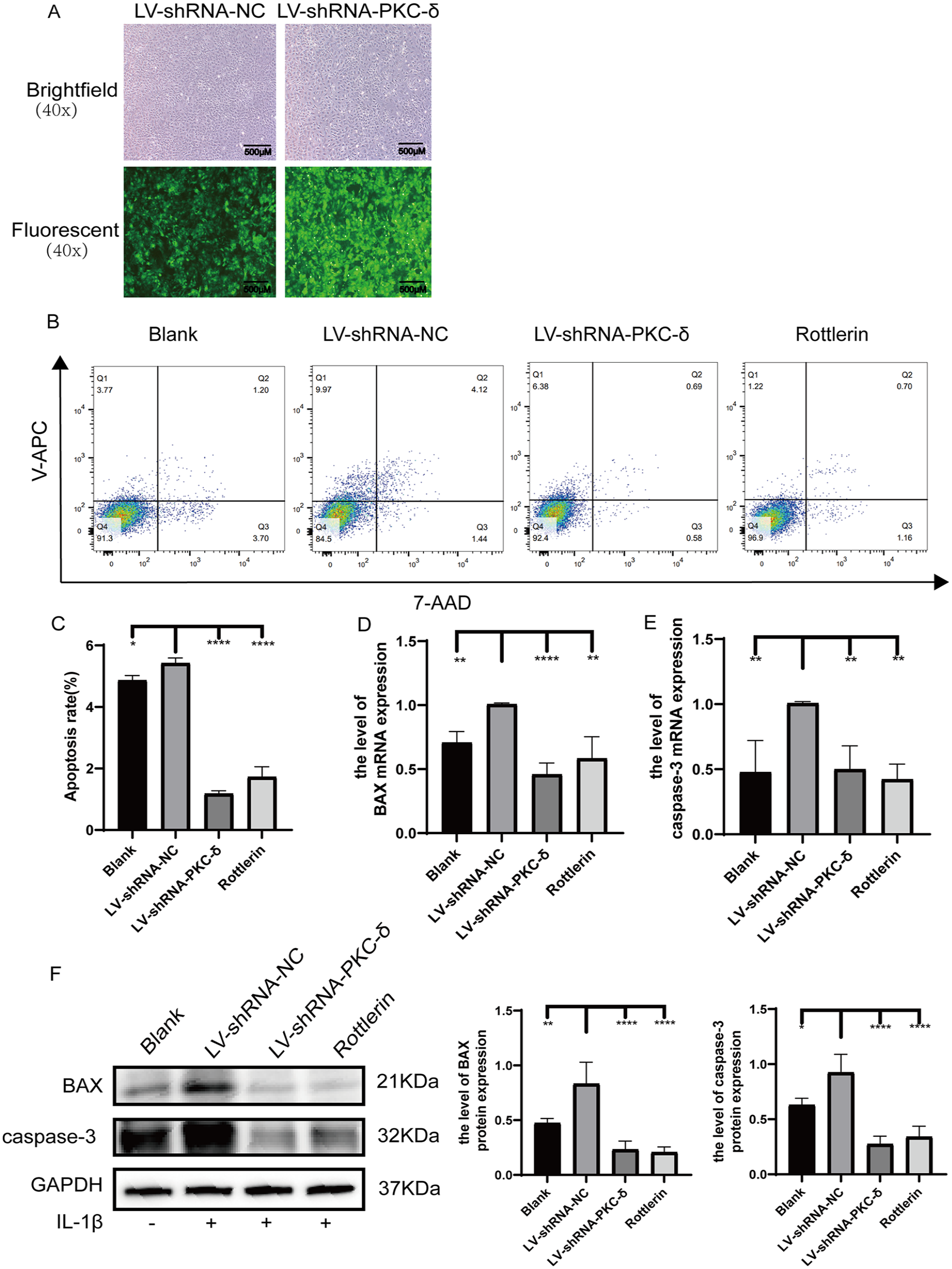
Apoptosis of chondrocytes in different groups of SD rats. (
Influence of PKC-δ on the JNK and P38 MAPK Signaling Pathway and Its Mechanism of Action
We assayed the mRNA exposure levels of PKC-δ in the chondrocytes of SD rats, a model of OA, after stimulation with IL-1β (10 ng/ml) via qRT-PCR. The mRNA exposure of PKC-δ was noticeably lower in the blank, LV-shRNA-PKC-δ, and rottlerin groups in comparison with the LV-shRNA-NC counterpart (
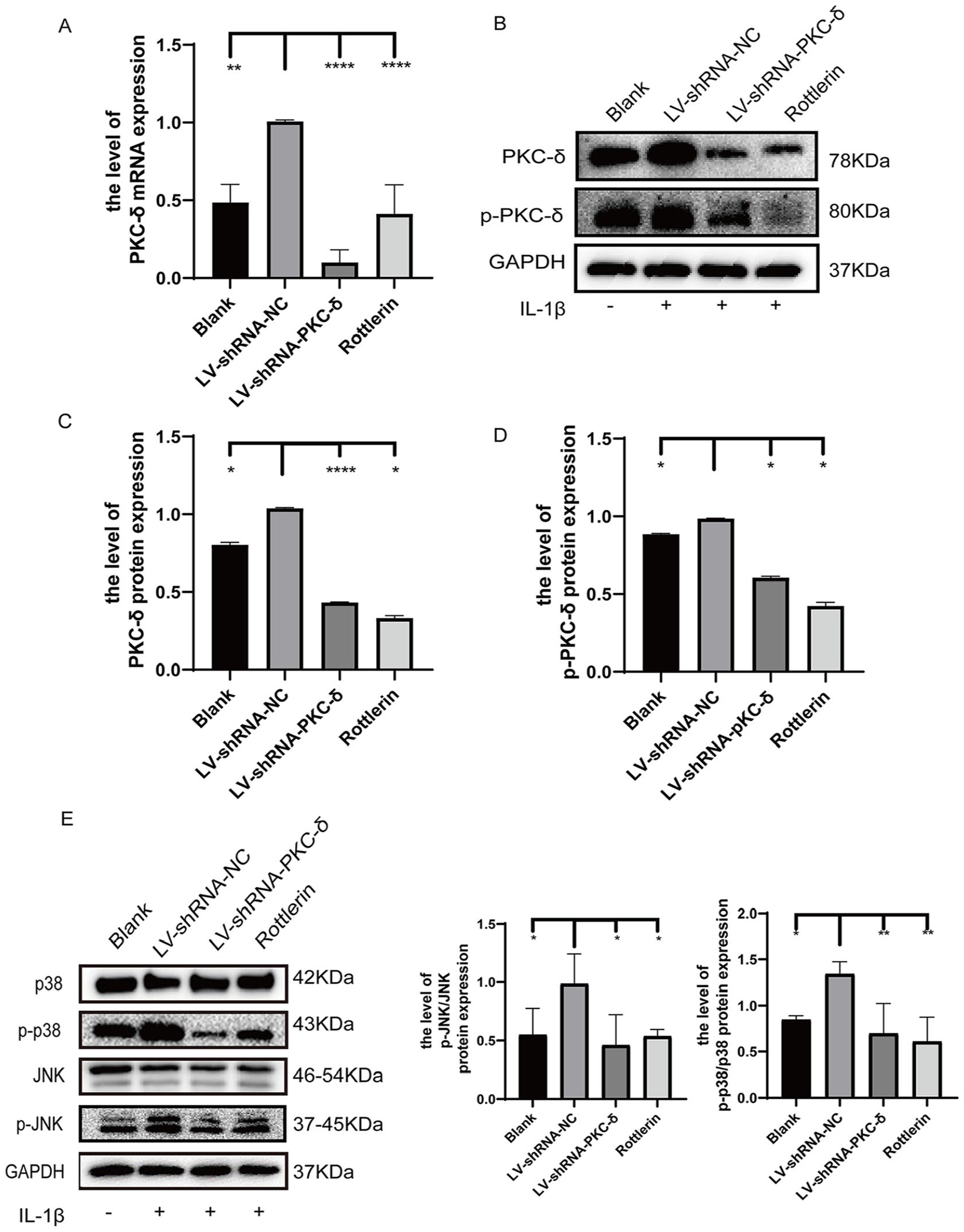
Effect of PKC-δ on the JNK and p38 MAPK signaling pathways and the mechanism involved. (
In summary, IL-1β induces PKC-δ expression and activation in chondrocytes, and the activated PKC-δ can promote the performance of BAX and caspase-3 apoptosis-related genes through regulation of JNK and p38 MAPK signaling cascades, thereby causing chondrocyte apoptosis and promoting OA progression.
Discussion
OA is the most prevalent chronic articular disorder in the elderly population and can lead to progressive destruction of articular cartilage, ultimately resulting in irreversible disability. The sole resident cell in articular cartilage, the chondrocyte, undergoes changes in proliferation, viability, secretion, and metabolism as OA develops, so damage to the function and survival of chondrocytes will lead to degeneration of articular cartilage. 37 Several studies confirmed that OA chondrocyte death has the morphological and molecular characteristics of apoptosis. 38 Apoptotic mechanisms play a critical role in cartilage degeneration, and apoptosis inhibition has been suggested as a means of protecting articular chondrocytes from OA.27,39,40 Although multiple proteins are involved in apoptosis, PKC-δ is pivotal as a signaling molecule within the apoptotic pathway; moreover, activated PKC-δ is a crucial mediator of apoptotic signaling. 41 PKC-δ has been reported to be a pro-apoptotic protein. In systemic lupus erythematosus and lymphoproliferative diseases, PKC-δ shows pro-apoptotic activity. 42 It also promotes apoptosis of renal cells in acute kidney injury. 43 Finally, activated PKC-δ promotes apoptosis in prostate cancer cells. 12 Nevertheless, the expression and activation of PKC-δ in OA and its relationship with chondrocyte apoptosis appear to be unexplored in domestic and international studies. In this research, immunohistochemical analysis indicated that the human osteoarthritic cartilage showed higher expression of PKC-δ and p38 proteins relative to normal cartilage. Moreover, TUNEL assays revealed a higher rate of apoptosis of cells from chondrocytes from osteoarthritic rather than from normal cartilage. Taken together, these findings confirm the ring-breaking function of PKC-δ in human OA. Thus, we recommend PKC-δ as a strategic potential target in OA therapy.
PKC-δ is broadly distributed in cells and tissues but is controlled by different molecular mechanisms. Activated by distinct mechanisms, PKC-δ plays diverse functions and critical positions in cellular processes such as apoptosis, polarization, and growth control. 44 Whether tested in vivo or in vitro, IL-1β, being one of the essential pro-inflammatory cytokines, leads to cartilage cell apoptosis, cartilage matrix degradation, and joint inflammation, thereby promoting OA progression.45,46 For this reason, a model of in vitro OA simulated by IL-1β was used in this study. Our present leads indicate that the mRNA of PKC-δ increases following IL-1β application to rat chondrocytes and the protein levels of both PKC-δ and p-PKC -δ increase in dose-dependent ways. Thus, the pro-inflammatory factor IL-1β can activate PKC-δ and play a role in its phosphorylation, a finding in line with those of previous studies. 13 In addition, we utilized flow cytometry to measure whether lentiviral silencing or the use of inhibitors followed by PKC-δ reduced chondrocyte apoptosis. The outcomes revealed that the proportion of chondrocytes in both early and late stages of apoptosis were markedly lower in the LV-shRNA-PKC-δ, rottlerin, and the blank groups in comparison with the LV-shRNA-NC counterpart. These findings indicate that chondrocyte apoptosis increases after PKC-δ activation by IL-1β, and that both silencing PKC-δ expression and adding rottlerin to inhibit phosphorylation of PKC-δ reduce chondrocyte apoptosis. This result is in line with that of Wang et al. 27 who found that chondroprotective effects on OA chondrocytes could be produced by suppressing apoptosis in chondrocytes cultivated with IL-1β. A growing body of research has also shown that members of the BCL-2 series (1) regulate chondrocyte viability and apoptosis and (2) initiate the activity of caspases in a cascade reaction. The activation of caspases is a core event in apoptosis in which the family causes cell destruction. 47 A corresponding increase in BAX levels and caspase-3 cleavage can promote apoptosis, and inhibition of BAX and caspase-3 activation may be critical for reducing chondrocyte apoptosis. 48 In ischemic stroke, chlorpromazine and promethazine exert neuroprotective effects by inhibiting the NOX/Akt/PKC pathway, reducing BAX and caspase-3, and upregulating BCL-XL, which are involved in the neuronal apoptosis mechanism. 49 The activation of PKC-δ and downregulation of some proto-oncogenes may lead to caspase-3 protein activation to induce apoptosis. 50 Consistent with previous studies, our work used qRT-PCR and Western Blot to detect these mediators of apoptosis. We found that silencing or inhibiting PKC-δ diminished the expression of BAX and caspase-3 in IL-1β-induced cells in chondrocytes. The present findings further suggest that PKC-δ exerts a pro-apoptotic effect upon chondrocytes by modulating critical mediators, including BAX and caspase-3.
Triggering the MAPK pathway can lead to the induction of apoptosis, and the MAPK pathway is one of the major upstream signaling pathways that cause apoptosis in chondrocytes.17,51 PKs perform tactical regulation by phosphorylating other target proteins and thus functionally regulate intracellular signal transduction pathways; consequently, any interference with their function may potentially lead to major downstream effects. 52 After cells are stimulated by other factors such as growth factors and inflammation, MAPK is activated by receiving activation signals and exhibits cascading phosphorylation. In mammals, the JNK and p38 MAPK signal pathways function critically during stress responses involving inflammation and apoptosis, as shown by Gonçalves et al. 53 who confirmed that activation of the PKC-δ/P38 MAPK pathway mediates podocyte apoptosis. Piao et al. 54 established that phosphorylation of JNK and PKC-δ participated in this upstream effect of inducing the apoptosis of U937 cells. It has not been explained, however, whether PKC-δ activation exerts its effect on regulating chondrocyte apoptosis in OA through phosphorylation of the JNK and p38 MAPK pathways. The p38 and JNK MAPK phosphorylation pathways were demonstrated to play critical parts in vascular dementia, with the anti-apoptotic protein BCL-2 being down-regulated and the pro-apoptotic genes BAX, caspase-3, and cleaved-parp being upregulated. 55 Single exercise triggers upregulation of p53 and caspase-3 as well as increased phosphorylation of p38 MAPK and JNK during neutrophil apoptosis. 56 Such studies imply that MAPK activation leads to phosphorylation of JNK and p38 MAPK, which, in turn, triggers transcription factors and stimulates apoptosis.
In this work, the mechanism and signaling pathway by which PKC-δ promotes apoptosis was further investigated by detecting the exposure levels for JNK and p38 and their phosphorylated proteins (p-JNK and p-p38) in IL-1β-induced osteoarthritic chondrocytes in vitro through Western blot. The outcomes revealed that both JNK and p38 protein presentation and phosphorylation levels in the MAPK pathway were decreased after silencing PKC-δ or inhibiting PKC-δ phosphorylation. We suggest that IL-1β activates PKC-δ, an event which then promotes apoptosis in OA chondrocytes by regulating phosphorylation along the JNK and p38 MAPK signal pathways, with upregulation of BAX and caspase-3 activity.
In summary, we demonstrated in an in vitro OA model that (1) IL-1β-induced PKC-δ expression and activation in chondrocytes and (2) activated PKC-δ can promote BAX and caspase-3 expression via regulation of the JNK and p38 MAPK signaling cascades, thereby causing chondrocyte apoptosis and promoting OA progression. Accordingly, both the PKC-δ signaling and the JNK and p38 MAPK pathways could be targets for the treatment of inflammation-induced OA. This study is anchored on the pro-chondrocyte apoptotic properties of PKC-δ under IL-1β induction ex vivo, which suggested to us that apoptosis inhibitors may become a new therapeutic option for OA patients. The damaging effects of PKC-δ in OA have not been tested in animals, and whether PKC-δ inhibitors can attenuate OA pathology still needs further validation. In the meantime, PKC-δ activators should also be tested in OA studies.
Supplemental Material
sj-pdf-1-car-10.1177_19476035231181446 – Supplemental material for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways
Supplemental material, sj-pdf-1-car-10.1177_19476035231181446 for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways by Jinfeng Lu, Miao Yu and Jia Li in CARTILAGE
Supplemental Material
sj-tif-3-car-10.1177_19476035231181446 – Supplemental material for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways
Supplemental material, sj-tif-3-car-10.1177_19476035231181446 for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways by Jinfeng Lu, Miao Yu and Jia Li in CARTILAGE
Supplemental Material
sj-xls-2-car-10.1177_19476035231181446 – Supplemental material for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways
Supplemental material, sj-xls-2-car-10.1177_19476035231181446 for PKC-δ Promotes IL-1β-Induced Apoptosis of Rat Chondrocytes and Via Activating JNK and P38 MAPK Pathways by Jinfeng Lu, Miao Yu and Jia Li in CARTILAGE
Footnotes
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study was supported by the National Natural Science Foundation of China (grant no.: 81760402) and “Medical Excellence Award” Funded by the Creative Research Development Grant from the First Affiliated Hospital of Guangxi Medical University.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
The Medical Ethics Committee of the First Affiliated Hospital of Guangxi Medical University approved this study (Approval number: 2016-KY-114).
Trial Registration
Not applicable.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.