
Abstract
Purpose:
To explore the relationship between insulin-like growth factor (IGF)-1R expression and the pathological progression of Kashin-Beck disease (KBD).
Design:
KBD cartilage samples were collected from 5 patients. Additionally, T-2 toxin was administered to rats fed a selenium (Se)-deficient diet, and their knee joints were collected. Human C28/I2 chondrocytes and mouse hypertrophic ATDC5 chondrocytes were cultured in vitro and treated with T-2 toxin and Se supplementation. Subsequently, the cultured human and mouse chondrocytes were treated with the IGF-1R inhibitor, picropodophyllin. Chondrocyte death and caspase-3 activity were analyzed using flow cytometry and a specific kit, respectively. Protein and mRNA expression levels of IGF-1R and matrix molecules were measured using immunohistochemistry, western blotting, and quantitative real-time reverse transcription-polymerase chain reaction analyses.
Results:
The cartilages from patients with KBD and T-2 toxin-treated rats on a Se-deficient diet showed significantly decreased expression of IGF-1R compared to cartilages from controls. T-2 toxin decreased IGF-1R mRNA and protein levels in both C28/I2 and hypertrophic ATDC5 chondrocytes in a dose-dependent manner; however, Se supplementation reduced the decrease of IGF-1R induced by T-2 toxin. Furthermore, inhibition of IGF-1R resulted in chondrocyte death of C28/I2 and hypertrophic ATDC5 chondrocytes, as well as decreased type II collagen expression and increased MMP-13 expression at the mRNA and protein levels.
Conclusion:
Downregulation of IGF-1R was associated with KBD cartilage destruction. Therefore, inhibition of IGF-1R may mediate chondrocyte death and extracellular matrix degeneration related to the pathological progression of KBD.
Keywords
Introduction
Kashin-Beck disease (KBD) is a chronic, endemic, deformable osteoarthropathy (OA) with a high rate of disability, which seriously endangers human health. T-2 mycotoxin contamination and selenium (Se) deficiency have been recognized as the main causes of KBD, which lead to cartilage damage.1,2 Continuous development of KBD cartilage destruction and lack of self-repair ability have been shown to progressively cause impaired mobility, disability, and chronic pain. 1 While KBD cartilage destruction was mainly related to cartilage matrix metabolism, degeneration, and chondrocyte death.1,3 Besides, chondrocyte necrosis in the deep zone and apoptosis in the middle zone of cartilage have been identified as the main pathological features of KBD,4,5 but the underlying pathogenic mechanism remains unclear.
Our research group has previously described the histopathology of chondronecrosis in the knee articular cartilage in a rat model of KBD (induced by T-2 toxin and Se-deficient conditions), which showed similar pathological changes as observed in children with KBD.6,7 Multiple studies have demonstrated that T-2 toxin promotes cartilage intracellular impairment and induces chondrocytes apoptosis. 8 Therefore, T-2 toxin has been shown to regulate levels of type II collagen (Col II) and its regulatory enzymes MMPs/tissue inhibitor of metalloproteinase-1 (TIMP-1) in this rat model of KBD. 9
Insulin-like growth factor-1 (IGF-1)/IGF-1R signaling affects hormones and mechanical signals through an autocrine/paracrine pathway, which enhances interactions between cells, thus promoting chondrocytes growth, survival, and differentiation.10,11 In IGF-1R knockout mice, the decreased bodyweight, growth retardation, and significantly increased chondrocytes apoptosis were observed. 12 These mice also displayed muscular hypoplasia and delayed ossification, indicating that IGF-1R plays an important role in growth and development. Moreover, phosphorylation of IGF-1R is known to activate downstream pathways that upregulate transcription of Col II and aggrecan, which promote extracellular matrix (ECM) synthesis. 13 The cartilage matrix protein, decorin, can also impact IGF-1R-dependent downstream signaling activation, thus affecting chondrocyte survival. 14
To further assess the role of IGF-1R in regulating chondrocyte growth, this study investigated the relationship between IGF-1R and cartilage destruction in the development of KBD pathogenesis. And targeted IGF-1R inhibition in chondrocytes was conducted to elucidate its role in regulating chondrocyte death.
Methods
Human Cartilage Tissues
The cartilage samples were acquired from 5 children patients with KBD who lived in diseased areas and died from accidents or other diseases. The adult cartilage samples were obtained from 5 KBD patients undergoing joint replacement surgery at the Xi’an Jiaotong University of Medicine from 2010 to 2014. All the patients were diagnosed according to the internationally recognized diagnostic criteria (GB16003-1995) for KBD in China. The normal cartilage samples were obtained from non-KBD area patients, who had died from traffic accidents or amputations resulted from trauma, with no history of any form of osteoarthritis or other inflammatory joint diseases. The samples were acquired with the informed consent of each patient or guardian and were approved by the Human and Ethical Committee for Medical Research at Xi’an Jiaotong University, School of Medicine (No. 3063058, Dr. Yong Liu, Director).
Experimental Animal Model
Nighty 1-month-old Sprague-Dawley rats (male, mass: 60-80 g) were purchased from the animal center of Xi’an Jiaotong University. The rats were randomly divided into a control group and a Se-deficient group that fed with normal diet or Se-deficient diet for 4 weeks, respectively. The normal diet was prepared based on the AIN-93G and AIN-93-VX contained 0.35 mg/kg sodium selenite (Na2O4Se), while the sodium selenite was omitted in the mineral mix in a Se-deficient diet. The groups were then subdivided into 4 subgroups as follows: normal group (n = 20), Se-deficient group (n = 20), T-2 toxin group (100 ng/g body weight [BW]/day; n = 25), and Se-deficient plus T-2 toxin group (100 ng/g BW/day; n = 25). The rats were continuously fed with each specific diet for another 4 weeks, and T-2 toxin was intragastrically administrated. The protocol was approved by the Animal Ethics Committee, Medical School of Xi’an Jiaotong University (No. 0074). The use of animals in this study was by following National Institute of Health publication 85-23, Guide for Care and Use of Laboratory Animals.
Cell Culture
The C28/I2 immortalized chondrocytes were a gift from Mary B. Goldring (Hospital for Special Surgery, New York, USA). The ATDC5 chondrogenic cells were obtained from the European Collection of Cell Cultures. C28/I2 cells and hypertrophic ATDC5 cells were cultured in the Dulbecco’s modified Eagle’s medium/F-12 medium (HyClone, Logan, UT) with 10% fetal bovine serum (FBS; Gibco, Carlsbad, CA) containing various concentration gradients of T-2 toxin (T002980, TRC, Canada) and/or plus Na2O4Se (0.1 μg/mL) for 72 hours and 48 hours, respectively. The ATDC5 chondrocytes were differentiated into hypertrophic chondrocytes by using a medium containing 1% ITS (insulin-transferrin-selenium mix; I3146, Sigma) for 21 days.
IGF-1R Inhibition
The IGF-1R signaling specific inhibitor, picropodophyllin (PPP; Selleck, Houston, TX), was used in C28/I2 and hypertrophic ATDC5 cells with various concentrations for a different time, and the cells were used for further experiments.
Immunohistochemical (IHC) Staining and Quantification
Sections of 5 µm thickness cut from paraffin-embedded tissue blocks were deparaffinized and rehydrated. Samples were performed with a primary antibody to IGF-1R (1: 50, Abcam, Cambridge, MA), and then incubated with horseradish peroxidase-conjugated secondary antibody. The 3,3′-diaminobenzidine was used to visualize the IHC reaction (DAB; Zhongshan Golden Bridge, Beijing, China). The histologic sections were photographed using an Olympus BX51 microscope equipped for digital image acquisition.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA was extracted using TRIzol reagent (Invitrogen, Carlsbad, CA). The complementary DNA was prepared from 1 mg of RNA using the RevertAid First-Strand Synthesis kit (Fermentas, Waltham, MA). The qRT-PCR reaction was performed on an iQ5 cycler (Biorad, Munich, Germany) using SYBR Green PCR Master Mix (Takara, Japan) with specific primers. The relative gene expression was normalized to the housekeeping gene (GAPDH) and quantified using the ΔCt method. The primer pairs for qRT-PCR are shown in the Supplemental Table 1.
Western Blotting
The protein samples separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis were transferred to polyvinylidene difluoride membranes (Millipore, Beverly, MA), and then incubated with IGF-1R antibody (1:1000, Abcam), MMP-13 (1:1000, Abcam), and Col II (1:500, Abcam). After incubated with anti-rabbit IgG (1:2000; CWBio, China), the membranes were developed with an enhanced chemiluminescence reagent (Millipore), and visualized by Chemidoc transilluminator (BioRad). Protein levels were standardized by comparison with GAPDH (1:1000; Proteintech).
Flow Cytometry Analysis
The chondrocyte apoptosis rate was analyzed using the Annexin V–FITC and PI apoptosis detection kit (BD Biosciences, San Diego, CA) and cells were double-stained with PI Annexin V and 7-amino-actinomycin. Samples were analyzed using a FACSCalibur flow cytometer (BD Biosciences).
Caspase-3 Activity Analysis
Caspase-3 activity was measured with a caspase-3 assay kit (Abcam, ab39401) according to the manufacturer’s protocol. And the caspase-3 activity was measured by fluorescence using a microplate reader at 405 nm.
Statistical Analysis
SPSS statistical software (Version 24.0; SPSS Inc., Chicago, IL) was used for data analysis, and the data were expressed as mean ± standard deviation (SD). T-test was used in the comparison between the 2 groups, and one-way analysis of variance (ANOVA) was used in the multigroups comparison. The LSD-q test was used for the further pairwise comparison. A P value of <0.05 was considered statistically significant.
Results
Decreased IGF-1R Immunostaining in Cartilages from Patients with KBD
The representative images of IHC showed that IGF-1R decreased in KBD children cartilage compared to that in healthy children cartilage ( Fig. 1A ), and the immunostaining quantitation results showed the lower ratio of IGF-1R-positive cells in the middle zone from KBD children cartilage than healthy cartilage, but there was no significant difference in the superficial zone and deep zone ( Fig. 1B ). Besides, IGF-1R expression was decreased in the KBD adult cartilage than in healthy adult cartilage ( Fig. 1C ). Meanwhile, the quantitation results showed a significant difference in superficial, middle, and deep zones of the articular cartilage from KBD adult cartilage compared to healthy adult cartilage ( Fig. 1D ).
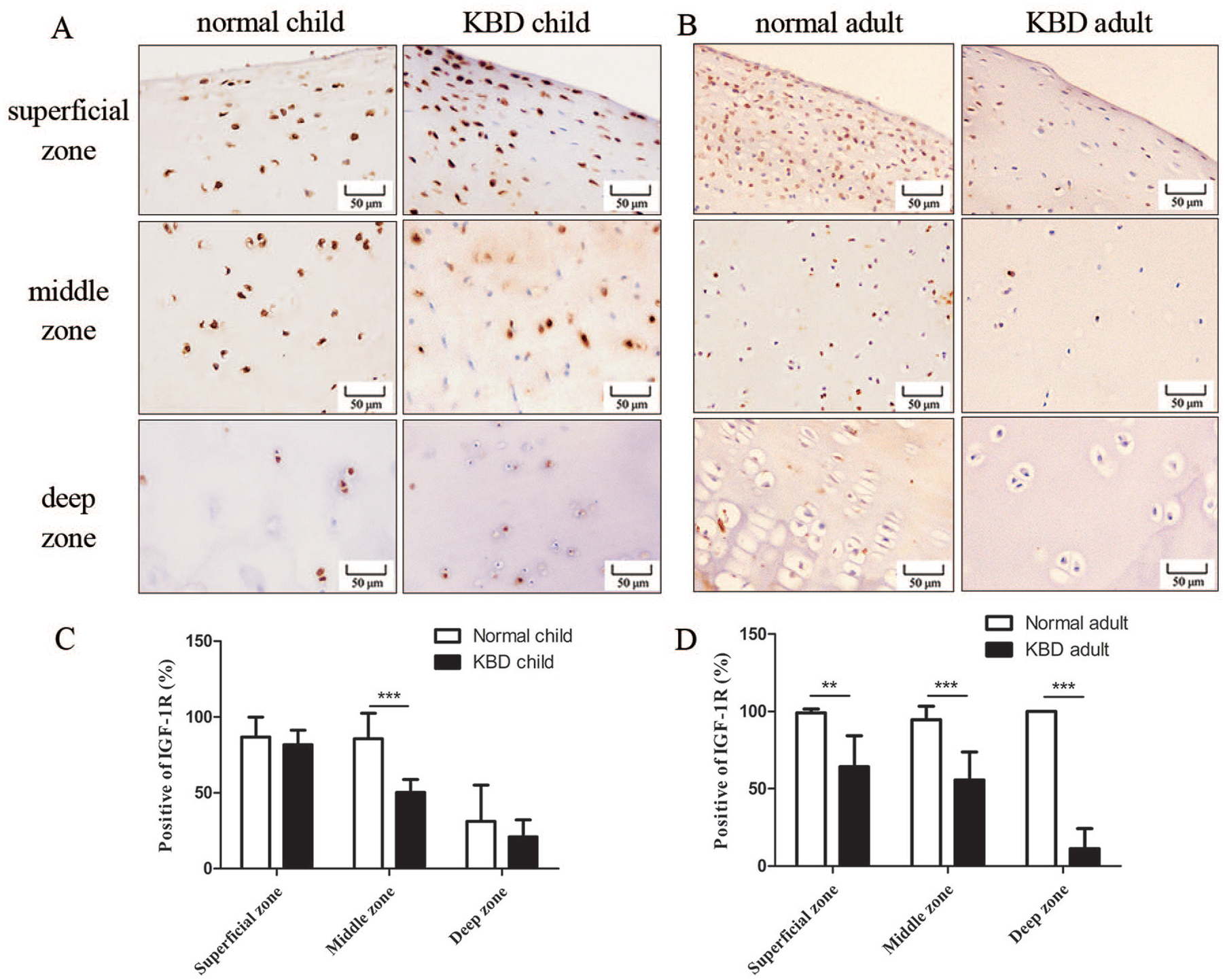
The immunostaining of IGF-1R in articular cartilage from healthy and KBD patients. (
Decreased IGF-1R Immunostaining in Cartilages of Rat Model of KBD
IGF-1R expression was examined using IHC staining in articular cartilage and epiphyseal plate of rats treated with T-2 toxin and fed a Se-deficient diet. The IGF-1R expression was decreased in articular cartilages from the Se-deficient, T-2 toxin, and T-2 toxin plus Se-deficient groups compared to the control group ( Fig. 2A ). Compared with the control group, the ratio of IGF-1R-positive cells was significantly reduced in articular cartilage from the experimental groups ( Fig. 2C ). All epiphyseal plates of studied mice were divided into resting, proliferative, and hypertrophic zones. The representative images showed that IGF-1R expression was decreased in the Se-deficient, T-2 toxin, and T-2 toxin plus Se-deficient groups, but no IGF-1R staining was detected in the resting and proliferative zones of 3 experimental groups ( Fig. 2B ). The ratio of IGF-1R-positive cells significantly decreased in the experimental groups than that in the control group ( Fig. 2D ).
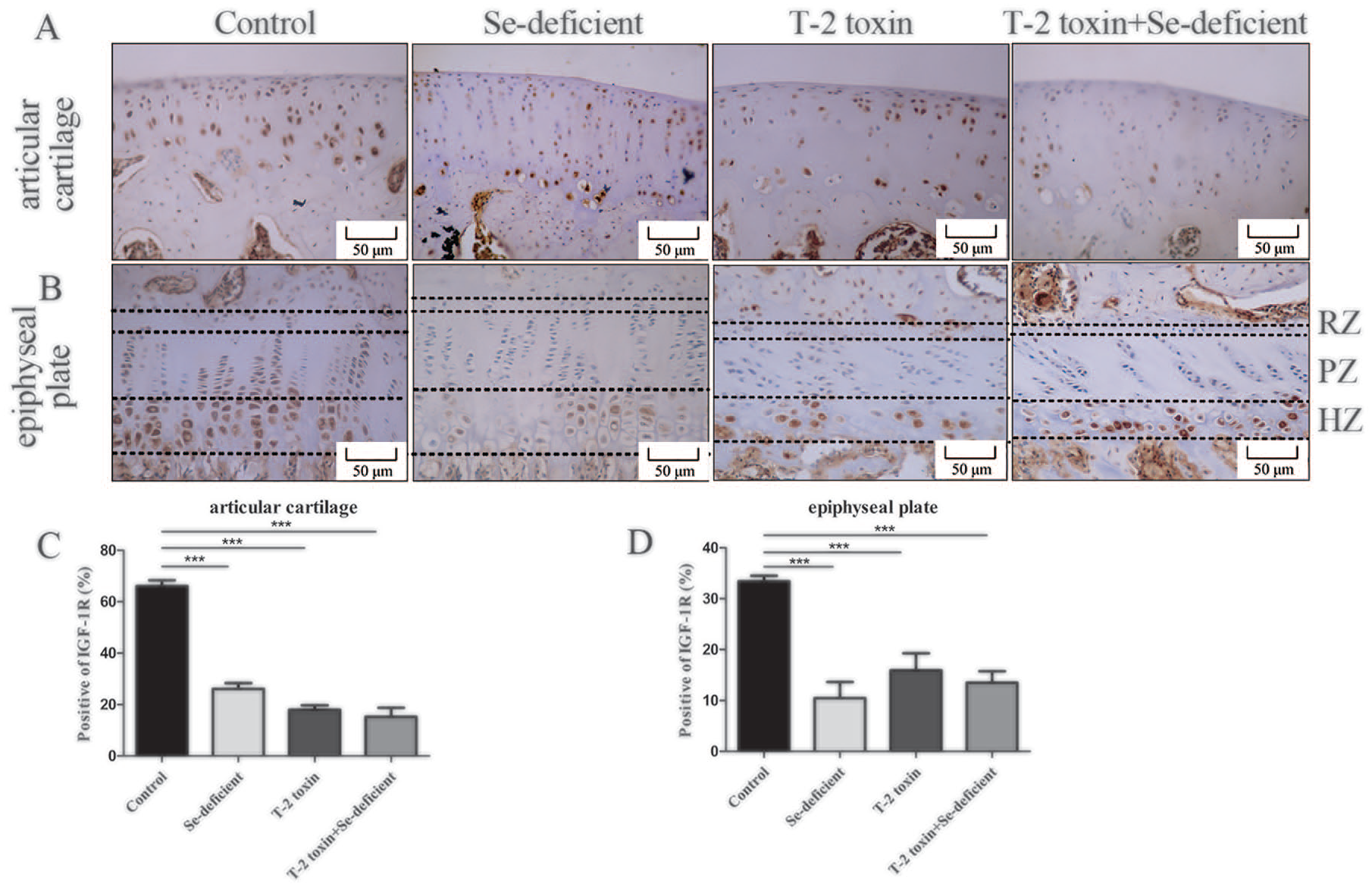
The immunostaining of IGF-1R in the cartilage of animal model. (
Effect of T-2 Toxin and Se Supplementation on IGF-1R Expression in Chondrocytes
Two types of chondrocytes were used to study the effect of T-2 toxin and Se on the expression of IGF-1R. In C28/I2 chondrocytes, the relative level of IGF-1R mRNA was decreased in a dose-dependent manner induced by T-2 toxin ( Fig. 3A ). Western blotting bands and cumulative optical density of IGF-1R demonstrated statistically lower protein levels in T-2 toxin-induced C28/I2 chondrocytes than that in the control group ( Fig. 3B and C ). Furthermore, the mRNA level of IGF-1R also showed a significant decrease in T-2 toxin-treated hypertrophic ATDC5 chondrocytes ( Fig. 3D ), as well as the protein level of IGF-1R was decreased, compared with the control group ( Fig. 3E and F ). Moreover, Se-supplementation in both C28/I2 and hypertrophic ATDC5 chondrocytes could partially block the T-2 toxin-induced decrease in IGF-1R expression.
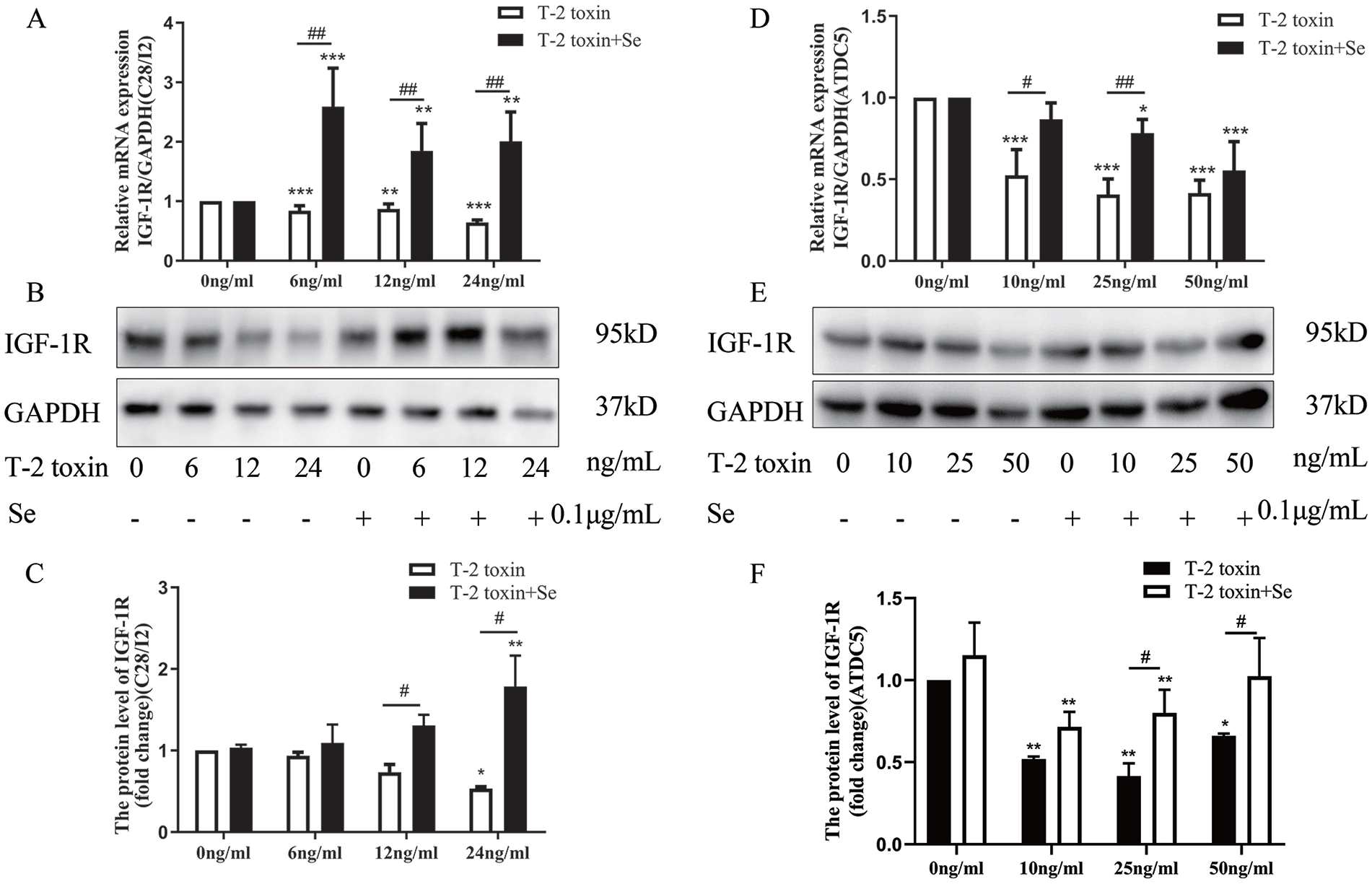
The effect of T-2 toxin and Se on the expression of IGF-1R in C28/I2 and hypertrophic ATDC5 chondrocytes. (
Effect of IGF-1R Inhibition on Chondrocyte Death
The effect of IGF-1R inhibition on chondrocyte death was detected by flow cytometry in 2 cell lines ( Fig. 4A and E ). The results showed that IGF-1R inhibition in C28/I2 chondrocytes dose-dependent increased chondrocyte apoptosis, but had no effect on chondrocyte necrosis at 24 hours ( Fig. 4B and C ). While both the chondrocyte apoptosis and necrosis were significantly increased in C28/I2 cells after 48 hours of IGF-1R inhibition ( Fig. 4B and C ). To further confirm the effect of IGF-1R on chondrocyte death, the caspase-3 activity was detected. The results showed that the caspase-3 activity increased at both 24 hours and 48 hours following PPP treatment in C28/I2 cells ( Fig. 4D ). In hypertrophic ATDC5 chondrocytes, the ratio of cell necrosis was increased, while no statistical difference in chondrocyte apoptosis was observed after 24 hours of IGF-1R inhibition ( Fig. 4F and G ). Moreover, after 48 hours IGF-1R inhibition, the ratio of chondrocytes apoptosis and necrosis both increased compared to that in controls ( Fig. 4F and G ). Besides, the caspase-3 activity was significantly increased in hypertrophic ATDC5 chondrocytes at 48 hours post-IGF-1R inhibition ( Fig. 4H ). The IGF-1R inhibitor solvent dimethyl sulfoxide had no effect on chondrocyte death at the maximum concentration.
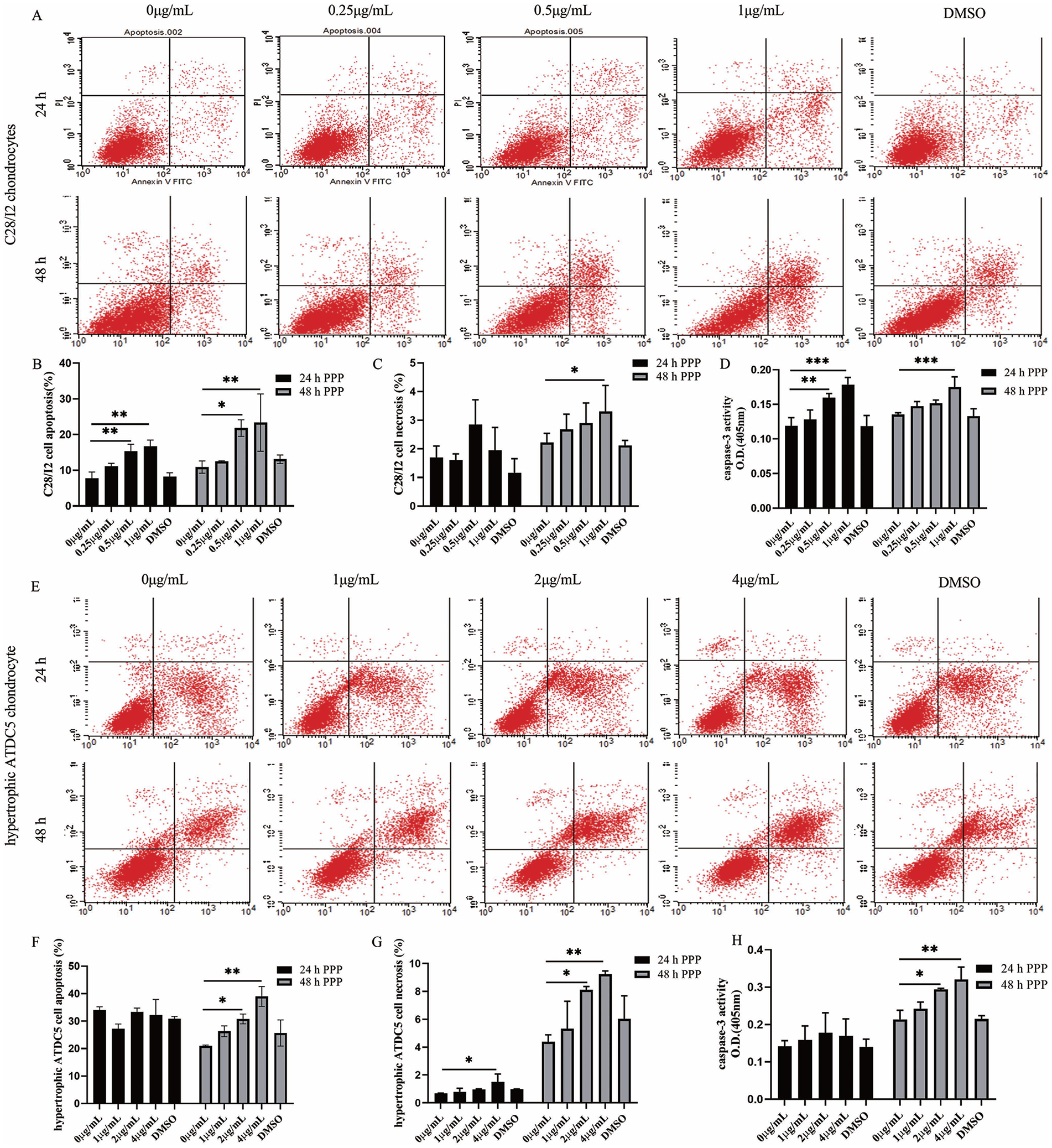
The effect of IGF-1R inhibition on chondrocyte death. (
Effect of IGF-1R on Cartilage Matrix Degradation
The cartilage matrix proteins were detected to investigate the effect of IGF-1R on matrix degradation. The result showed IGF-1R inhibition dose-dependently decreases Col II expression at the mRNA and protein levels ( Fig. 5A and B ), while the mRNA and protein expression of MMP-13 was increased in C28/I2 chondrocytes ( Fig. 5C and D ). Similar expression patterns were also observed in hypertrophic ATDC5 chondrocytes that the mRNA and protein expression of Col II was significantly decreased ( Fig. 5E and F ), while the expression of MMP-13 was increased after IGF-1R inhibition ( Fig. 5G and H ).
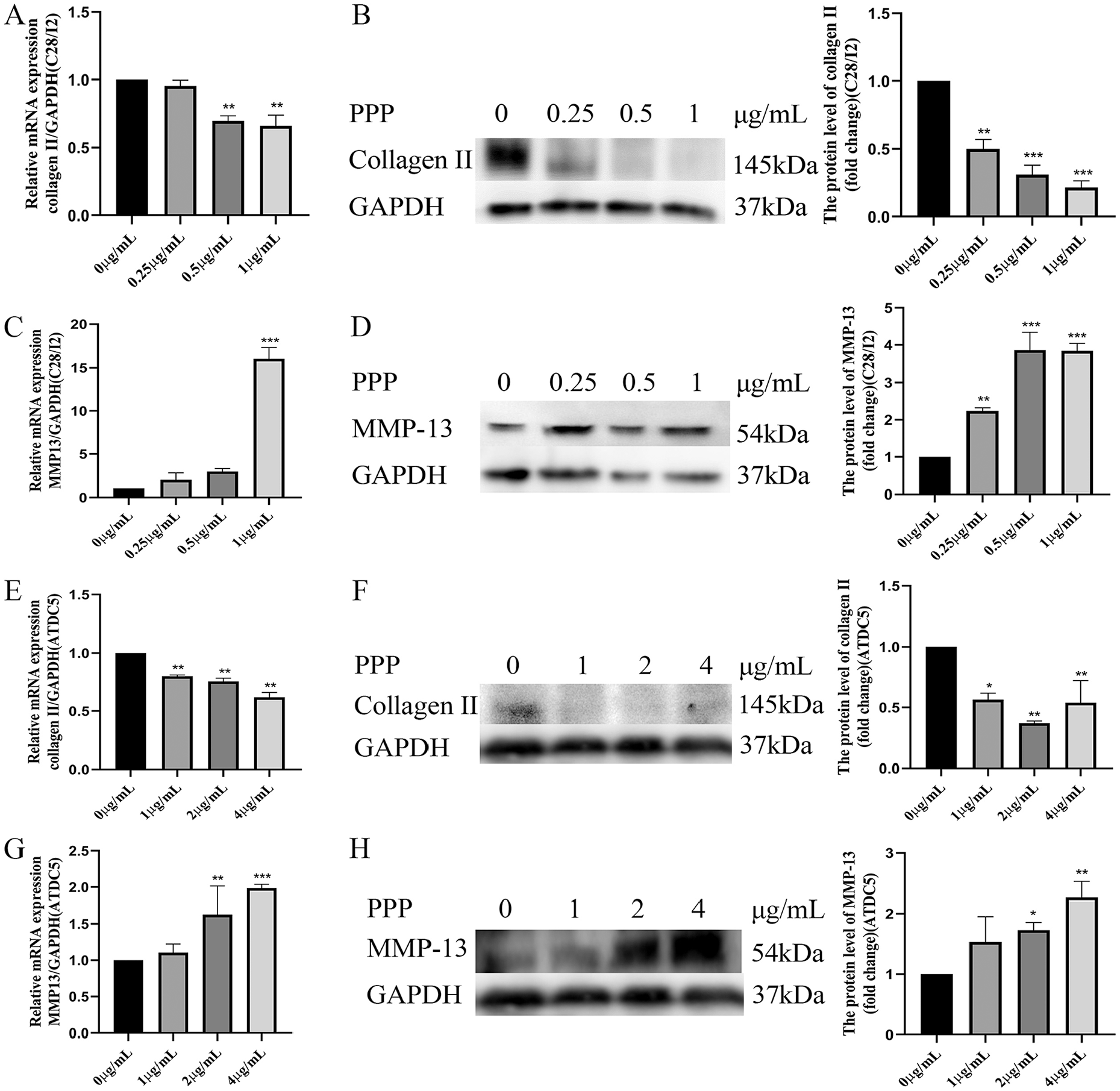
The effect of IGF-1R inhibition on the expression of Col II and MMP-13 in chondrocytes. (
Discussion
The main mechanisms of KBD cartilage destruction are excessive chondrocytes death and cartilage matrix degradation. This study showed a significant decrease in IGF-1R expression in the articular cartilage of patients with KBD, suggesting a correlation between abnormal IGF-1R expression and KBD cartilage destruction.
To further investigate the role of IGF-1R in KBD cartilage destruction, the T-2 toxin/Se-deficient rat model of KBD was used in this study. In previous studies, both T-2 toxin and Se deficiency led to cartilage destruction in rats, as well as reduced proteoglycan and Col II expression and altered expression of MMPs. 9 Besides, chondrocytes apoptosis was demonstrated by the presence of cell lacunae and increased expression of caspase-3, p53, and Bax in the rat cartilages.6,7 These chondrogenesis lesions were comparable to cartilage damage observed in KBD. Besides, cartilage-specific IGF-1R knock-out mice die shortly after birth and with less mineralized skeletons, as a result of severe chondrocyte defects characterized by reduced proliferation, delayed differentiation and hypertrophy, and increased chondrocyte apoptosis.11,12,15 Thus, decreased IGF-1R expression in articular cartilage and epiphyseal plate of rats exposed to T-2 toxin and Se-deficient conditions, suggesting that IGF-1R may involve in cartilage destruction caused by T-2 toxin in rats model. Articular cartilage has a highly organized structure composed of the superficial zone, middle zone, deep zone, and calcified zone. The chondrocyte phenotype and shape vary among the different zones. In this study, the subtle zonal differences of IGF-1R expression in rat articular cartilage may indicate that T-2 toxin has a different sensitivity to chondrocytes with different phenotypes, which need to be studied further.
Besides, the hypertrophic ATDC5 chondrocytes were used as targeted cells to investigate chondrocytes changes in the deep zone of articular cartilage where they act as the most commonly affected anatomical site of KBD patients. 1 T-2 toxin was shown to induce chondrocyte death by increasing the Bax/Bcl-2 ratio and enhancing matrix degradation through Col II and MMPs. 8 The decreased IGF-1R expression induced by the T-2 toxin indicates the IGF-1R may involve in the T-2 toxin-induced chondrocyte death. Moreover, Se as an essential nutrient and trace element that is required for human health, its supplementation partially increases the expression of IGF-1R, which also protects the cell from death. Together, the results from the KBD rat model and chondrocytes indicate the involvement of IGF-1R in the T-2 toxin-induced cartilage and chondrocyte destruction.
IGF-1R/IGF-1 signaling activates critical cell survival factors to protect cells from apoptosis through several downstream pathways, including the phosphatidylinositol-3 kinase (PI3K)/protein kinase B (PKB) pathway, interleukin-Iβ-converting enzymes (ICE), and ICE-like proteases.16,17 A specific IGF-1R inhibitor PPP, which efficiently blocks IGF-1R activity, 18 was used to investigate the involvement of IGF-1R in regulating chondrocyte death and ECM degradation. Present data showed that the inhibition of IGF-1R induced both chondrocytes apoptosis and necrosis at 48 hours in these 2 cell lines. But the 24 hours IGF-1R inhibition caused apoptosis in C28/I2 and necrosis in hypertrophic ATDC5 cells, which is consistent with the KBD cartilage destruction that apoptosis occurs at the middle zone while necrosis mainly occurs at the deep zone of cartilage.3,5 Besides, a previous study showed that T-2 toxin increases apoptosis, with no effect on necrosis, of C28/I2 chondrocytes. 19 Several studies demonstrated the IGF-1R signaling regulating the proliferation and differentiation, which may speculate the role of IGF-1R in regulating the death of chondrocytes with different phenotypes in different ways. 16 Besides, the increased caspase-3 activity in chondrocytes after IGF-1R inhibition further confirming the involvement of IGF-1R in regulating chondrocyte death. Hence, IGF-1R inhibition in chondrocytes suggests its involvement in regulating KBD chondrocyte death.
Since IGF-1/IGF-1R signaling pathway also responsible for regulating ECM protein synthesis in articular cartilage, notably collagens and proteoglycan. 20 And gene silencing of IGF-1R downregulated Col II expression and upregulated MMPs in several cell types through activation of downstream PI3K signal pathways.13,20 Besides, the increased matrix metabolism during KBD cartilage destruction has been confirmed. Our present data showed that inhibition of IGF-1R in chondrocytes induced the downregulation of Col II and upregulation of MMP-13, indicating the involvement of IGF-1R in T-2 toxin-induced ECM degradation.
Taken together, this study showed decreased IGF-1R expression in the cartilage of KBD patients. Furthermore, T-2 toxin-induced the downregulation of IGF-1R in the cartilage of a KBD rat model and in chondrocytes in vitro. Inhibition of IGF-1R signaling was shown to result in chondrocyte death and ECM degeneration. Therefore, we speculate that downregulation of IGF-1R is involved in the development of KBD through regulation of chondrocyte death and ECM degradation. Further study is needed to clarify the specific mechanism of IGF-1R in the pathological progression of KBD cartilage destruction.
Supplemental Material
sj-pdf-1-car-10.1177_19476035211021890 – Supplemental material for Downregulation of Insulin-Like Growth Factor-1 Receptor Mediates Chondrocyte Death and Matrix Degradation in Kashin-Beck Disease
Supplemental material, sj-pdf-1-car-10.1177_19476035211021890 for Downregulation of Insulin-Like Growth Factor-1 Receptor Mediates Chondrocyte Death and Matrix Degradation in Kashin-Beck Disease by Meng Zhang, Wenjun Wang, Hui Wang, Yinan Liu, Zhengzheng Li, Chengfen Yi, Yawen Shi, Tianyou Ma and Jinghong Chen in CARTILAGE
Footnotes
Acknowledgments and Funding
The author(s) disclosed receipt of the following financial support for the research, authorship, and/or publication of this article: This study supported by the National Natural Science Foundation of China (No. 81872565, No. 81573102).
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Approval
The study was approved by the Human and Ethical Committee for Medical Research at Xi’an Jiaotong University, School of Medicine (No. 3063058, Dr. Yong Liu, Director).
Informed Consent
The samples were acquired with the informed consent of each patient or guardian.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.