
Abstract
Objective
Osteoarthritis (OA) is a common joint disorder, accompanied by extracellular matrix (ECM) degradation. Reportedly, long noncoding RNAs (lncRNAs) are involved in OA pathogenesis. However, the role of lncRNA FYVE, RhoGEF, and PH domain containing 5 antisense RNA 1 (FGD5-AS1) in OA development is still not fully clarified. This study was aimed to clarify the role of FGD5-AS1 in OA.
Methods
FGD5-AS1 and miR-302d-3p expression levels were determined in cartilage tissues and chondrocytes by quantitative real-time polymerase chain reaction (qRT-PCR). Chondrocytes (C20/A4 cells) were stimulated with interleukin 1β (IL-1β) to mimic the inflammatory environment of OA. Cell viability was detected by cell counting kit-8 and 5-ethynyl-2′-deoxyuridine assays. Cell apoptosis was measured by the caspase-3 activity assay and flow cytometry. Transforming growth factor beta receptors II (TGFBR2), matrix metalloproteinase 13 (MMP-13), and ADAM metallopeptidase with thrombospondin type 1 motif 5 expression levels were examined by qRT-PCR or Western blot. The regulatory relationships among FGD5-AS1, miR-302d-3p, and TGFBR2 were predicted by the StarBase v2.0, miRanda, miRDB, and TargetScan databases, and confirmed by dual-luciferase reporter assay and RNA immunoprecipitation assay.
Results
FGD5-AS1 and TGFBR2 expression levels were downregulated while miR-302d-3p expression was increased in cartilage tissues of OA patients. Knocking down FGD5-AS1 inhibited the viability of C20/A4 cells but induced apoptosis and ECM degradation, while FGD5-AS1 overexpression exerted opposite effects. MiR-302d-3p was identified as a target of FGD5-AS1, and TGFBR2 was identified as a target of miR-302d-3p. FGD5-AS1 positively regulated TGFBR2 expression by repressing miR-302d-3p expression, and miR-302d-3p inhibition or TGFBR2 restoration reversed the changes of cell viability, apoptosis, and ECM degradation induced by FGD5-AS1 knockdown.
Conclusion
FGD5-AS1 can probably inhibit OA progression by regulating miR-302d-3p/TGFBR2 axis.
Introduction
Osteoarthritis (OA), a chronic degenerative joint disease, is a common disease among old people and features with articular cartilage degradation and joint pain. 1 Multiple risk factors (such as aging, genetic factors, and biochemical and biomechanical imbalance) are involved in OA pathogenesis.2,3 During the OA pathogenesis, imbalanced anabolism and catabolism of the extracellular matrix (ECM) aggravate the lesion of cartilage and promote the disease progression.4,5 During this process, matrix metalloproteinase 13 (MMP-13) and ADAM metallopeptidase with thrombospondin type 1 motif 5 (ADAMTS5) are associated with the loss of type II collagen/aggrecan, which accelerate ECM degradation. 6 Besides, OA is considered as an inflammatory disease, and the injury of chondrocytes mediated by pro-inflammatory factors and oxidative stress also exacerbates the disease. 7
Accumulating evidence reveals that the OA progression is affected by the dysregulation of long noncoding RNAs (lncRNAs). For instance, lncRNA FOXD2-AS1 induces the proliferation of chondrocytes and promotes inflammation and ECM degradation, which facilitates OA progression 7 ; lncRNA MEG3 facilitates chondrocytes’ proliferation, and inhibits cell apoptosis and ECM degradation. 8 LncRNA FYVE, RhoGEF, and PH domain containing 5 antisense RNA 1 (FGD5-AS1) is a regulator in several cancers, including esophageal squamous cell carcinoma, 9 non–small cell lung cancer, 10 and oral cancer. 11 Moreover, FGD5-AS1 expression is reported to be downregulated in the gingival tissues of patients with chronic periodontitis, and FGD5-AS1 overexpression alleviates the lipopolysaccharide (LPS)-induced periodontitis via attenuating the inflammatory response. 12 However, to date, the role of FGD5-AS1 in OA progression still awaits more verification.
In this work, we mainly focused on the function of FGD5-AS1 in regulating ECM degradation and the viability of chondrocytes in inflammatory environment. We demonstrated that FGD5-AS1 could work as a competitive endogenous RNA (ceRNA) to modulate miR-302d-3p and transforming growth factor beta receptor 2 (TGFBR2), protecting chondrocytes from the injury induced by inflammation.
Materials and Methods
Tissue Specimens
All procedures in this study were approved by the Ethics Committee of Renmin Hospital of Wuhan University and performed according to the Declaration of Helsinki. The articular cartilages (n = 35) subjected to total knee replacement were obtained from OA patients (n = 35) in the Renmin Hospital of Wuhan University from March 2013 to July 2019, and the normal articular cartilage tissues (n = 35) from 35 patients without OA (who received amputation) were used as the control. All participants provided the written informed consent. The status of the patients was evaluated by Knee Injury and Osteoarthritis Outcome Score (KOOS) survey. Supplementary Table 1 shows the clinical characteristics of OA patients. The tissue samples were obtained during the surgery and then stored in liquid nitrogen at −196°C until the RNA extraction.
Cell Culture and IL-1β Stimulation
ATCC (American Type Culture Collection, Manassas, VA) was the provider of human chondrocytes cell line C20/A4. The cells were cultured in Dulbecco’s Modified Eagle’s Medium (DMEM; Gibco, Rockville, MD)/F12 supplemented with 10% fetal bovine serum (FBS; Invitrogen, Waltham, MA) and L-glutamine (Sigma-Aldrich, Louis, MO). The cells were cultured in an incubator containing 5% CO2 at 37°C. Interleukin-1β (IL-1β; Sigma-Aldrich, Louis, MO) was dissolved in double-distilled H2O according to the manufacturer’s instructions, with a storage concentration of 5 mg/mL. Then, IL-1β was diluted to 1, 5, or 10 µg/mL using serum-free DMEM/F12 medium, respectively. In this study, C20/A4 cells were treated with 1, 5, or 10 µg/mL IL-1β, respectively, for 48 hours to induce the inflammatory injury.
Cell Transfection
Overexpression plasmids for FGD5-AS1 and TGFBR2 were provided by Invitrogen Co. Ltd. (Carlsbad, CA). siRNAs were purchased from Ribobio (Guangzhou, China), and miRNA mimics and inhibitors were synthesized by Genepharma Co. Ltd. (Shanghai, China). C20/A4 cells were transfected with Lipofectamine 3000 (Invitrogen, Carlsbad, CA) according to the manufacturer’s instructions. Forty-eight hours later, chondrocytes were then treated with IL-1β (10 ng/mL) to induce the inflammatory injury.
Quantitative Real-Time Polymerase Chain Reaction (qRT-PCR)
Total RNA extraction from tissues or cells was performed by TRIzol reagent (Invitrogen, Carlsbad, CA), and then the total RNA was reversely transcribed into cDNA with PrimeScript RT Master Mix (for gene amplification; Takara, Dalian, China) or Mir-X miRNA First-Strand Synthesis Kit (for miRNA amplification; Takara, Dalian, China). With cDNA as template, SYBR Premix Ex Taq kit (Takara, Dalian, China) was used for qRT-PCR, which was performed on Applied Biosystems 7500 Fast System (ABI, Thermo Fisher Scientific, Inc., Foster City, CA). Glyceraldehyde 3-phosphate dehydrogenase (GAPDH) and U6 were considered as endogenous controls. The primers’ sequences are shown in Supplementary Table 2.
Cell Counting Kit-8 (CCK-8) Assay
C20/A4 cells in the logarithmic growth phase were trypsinized with trypsin, immersed in PBS, and centrifuged, and collected cell pellets were resuspended and the cells were counted. Next, the cells were transferred in 96-well plates (2 × 103 cells/well). At 0, 24, 48, 72, and 96 hours, 10 µL of CCK-8 solution (Dojindo Molecular Technologies, Tokyo, Japan) was loaded in each well and the cells were incubated for 2 hours. Then, the optical density values (OD450) of the cells were measured to evaluate the cell viability of C20/A4 cells.
Caspase-3 Activity Assay
The caspase-3 activity was assessed by caspase-3 colorimetric assay kit (Abcam, Cambridge, UK) in line with the manufacturer’s instruction. Briefly, the lysates of the transfected cells were centrifuged, and the supernatant was collected and then incubated with reaction buffer supplemented with caspase substrates at 37°C for 4 hours. The caspase-3 activity was evaluated by the absorbance at 405 nm through a microplate reader.
Flow Cytometry Analysis
Fluorescein isothiocyanate (FITC)-Annexin V/propidium iodide (PI) Apoptosis Detection kit (Thermo Fisher Scientific, Rockford, IL) was used to detect the apoptosis of C20/A4 cells. To be specific, in each group, about 1 × 105 transfected C20/A4 cells were rinsed with PBS, trypsinized, and resuspended in 100 µL of binding buffer. Then the C20/A4 cells were incubated with 5 µL of FITC-Annexin V staining solution and 5 µL of PI staining solution, respectively. Ultimately, the apoptosis of C20/A4 cells was detected by a flow cytometer (BD Biosciences, San Diego, CA).
Western Blot
Total proteins from cells were isolated with RIPA buffer (Beyotime, Shanghai, China), and the supernatant was collected after the centrifugation, and the protein concentration in the supernatant was detected with BCA Protein Assay kit (Thermo Fisher Scientific, Inc., Rockford, IL). After sodium dodecyl sulfate-polyacrylamide gel electrophoresis, the proteins were transferred onto the polyvinylidene fluoride (PVDF) membranes (Millipore, Billerica, MA). Following that, the PVDF membranes were blocked with 5% skim milk for 2 hours at room temperature and then incubated with the primary antibodies against TGFBR2 (1:1000, ab186838, Abcam, Cambridge, UK), MMP-13 (1:500, ab39012, Abcam), ADAMTS5 (1:1000, ab41037, Abcam), and β-actin (1:2000, ab8227, Abcam) at 4°C overnight. After being washed with TBST buffer, the membranes were incubated with the horseradish peroxidase-conjugated secondary antibodies (Beyotime, Shanghai, China) for 1 hour at room temperature. Finally, the protein bands were detected with ECL reagents (Amersham Biosciences, Pittsburg, PA).
5-Ethynyl-2′-Deoxyuridine (EdU) Assay
The proliferation of chondrocytes was assessed by EdU assay adopting Cell-Light EdU Apollo488 In Vitro Imaging Kit (RioBio, Guangdong, China) according to the manufacturer’s instruction. In brief, C20/A4 cells were incubated with EdU solution for 2 hours, and then the medium was discarded. Subsequently, the cells were fixed with 4% paraformaldehyde before the Apollo488 solution was used to mark the proliferative cells, and the nuclei of the cells were marked with Hoechst staining solution. Ultimately, the cells were observed under a fluorescent microscope (Olympus, Tokyo, Japan).
Bioinformatics and Dual-Luciferase Reporter Assay
Starbase 2.0 databases (http://starbase.sysu.edu.cn/) indicated that there were potential binding sites between FGD5-AS1 and miR-302d-3p. Three bioinformatics databases (miRanda, miRDB, and TargetScan) showed that there were putative binding sites between miR-302d-3p and TGFBR2. The wild-type luciferase reporter vectors were established by subcloning the PCR-amplified fragments of FGD5-AS1 and TGFBR2 3′-untranslated region (3′UTR) into the pmirGLO vectors (Promega, Madison, WI). Similarly, the mutant luciferase reporter vectors were obtained in the same way. C20/A4 cells were co-transfected with the reporter vectors and miR-302d-3p mimics or miR-302d-3p inhibitors. Forty-eight hours later, the relative luciferase activities of the cells in each group were assessed by the Dual-Luciferase Reporter Assay Kit (Promega, Madison, WI) according to the manufacturer’s instructions.
RNA Immunoprecipitation (RIP) Assay
EZ-Magna RIP kit (Millipore, Bedford, MA) was used to validate the direct interaction between FGD5-AS1 and miR-302d-3p. Briefly, C20/A4 cells were lysed in RIP buffer containing magnetic beads coupled with antibodies against Ago2 or IgG (negative control). The lysates were incubated for 6 hours at 4°C. Next, the mixtures were incubated with proteinase K to remove the proteins. Next, the RNA in immunoprecipitate was extracted with TRIzol method, and qRT-PCR was performed to detect FGD5-AS1 and miR-302d-3p expression levels.
Statistical Analysis
All experiments were performed in triplicate, and repeated for at least 3 times. SPSS 19.0 (SPSS Inc., Chicago, IL) was used to analyze the experimental data. All the data were presented as mean ± standard deviation (x ± s). Student’s t test was adopted for the comparison between 2 groups, and one-way analysis of variance (ANOVA) was used for the comparison among 3 or more groups. The correlation between 2 variables was evaluated by Pearson’s correlation coefficient. P < 0.05 has statistical significance.
Results
FGD5-AS1 Knockdown Repressed the Viability and Promoted the Apoptosis of C20/A4 Cells
First, qRT-PCR depicted that FGD5-AS1 expression was decreased in the OA cartilage tissues compared with normal cartilage tissues (
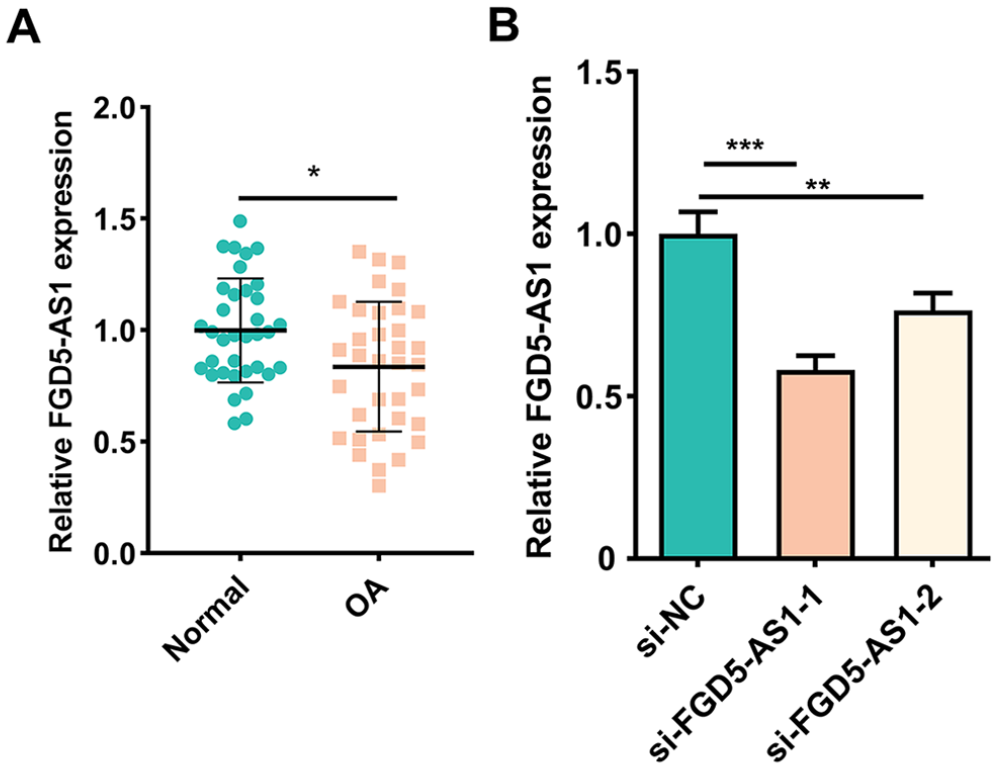
FGD5-AS1 expression in OA and normal cartilage tissues. (
FGD5-AS1 Overexpression Ameliorated the Injury of C20/A4 Cells Induced by IL-1β
Next, C20/A4 cells were treated with IL-1β to mimic the inflammatory environment in OA, and then we investigated the effects of FGD5-AS1. As shown, FGD5-AS1 expression was significantly elevated in C20/A4 cells transfected with pcDNA-FGD5-AS1 than that in the control group (
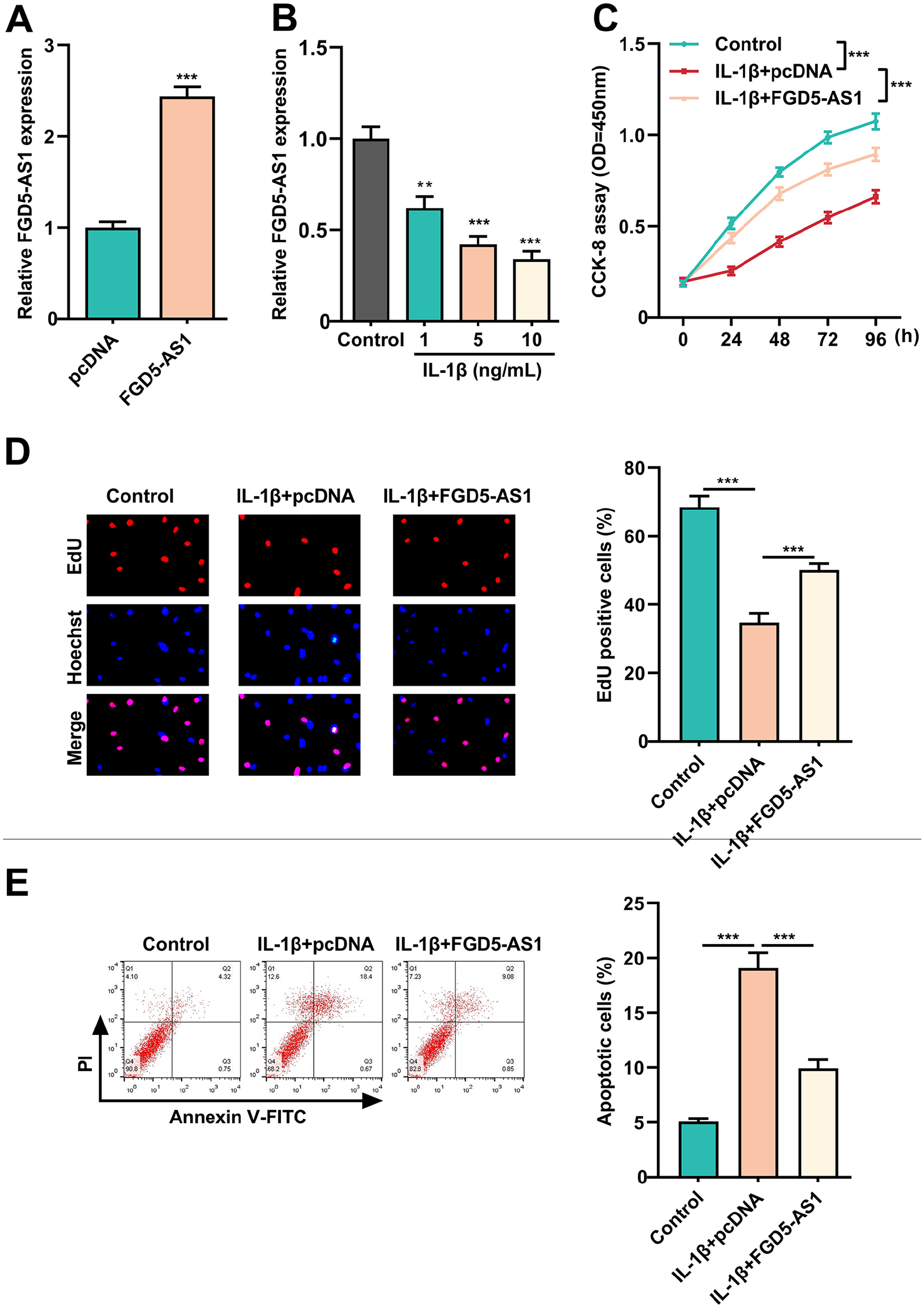
Impacts of FGD5-AS1 overexpression on chondrocyte proliferation, apoptosis, and ECM degradation after IL-1β treatment. (
FGD5-AS1 Sponged MiR-302d-3p and Repressed MiR-302d-3p Expression in Chondrocytes
MiR-302d-3p was among the candidate miRNAs that were predicted by Starbase 2.0 databases (http://starbase.sysu.edu.cn/;
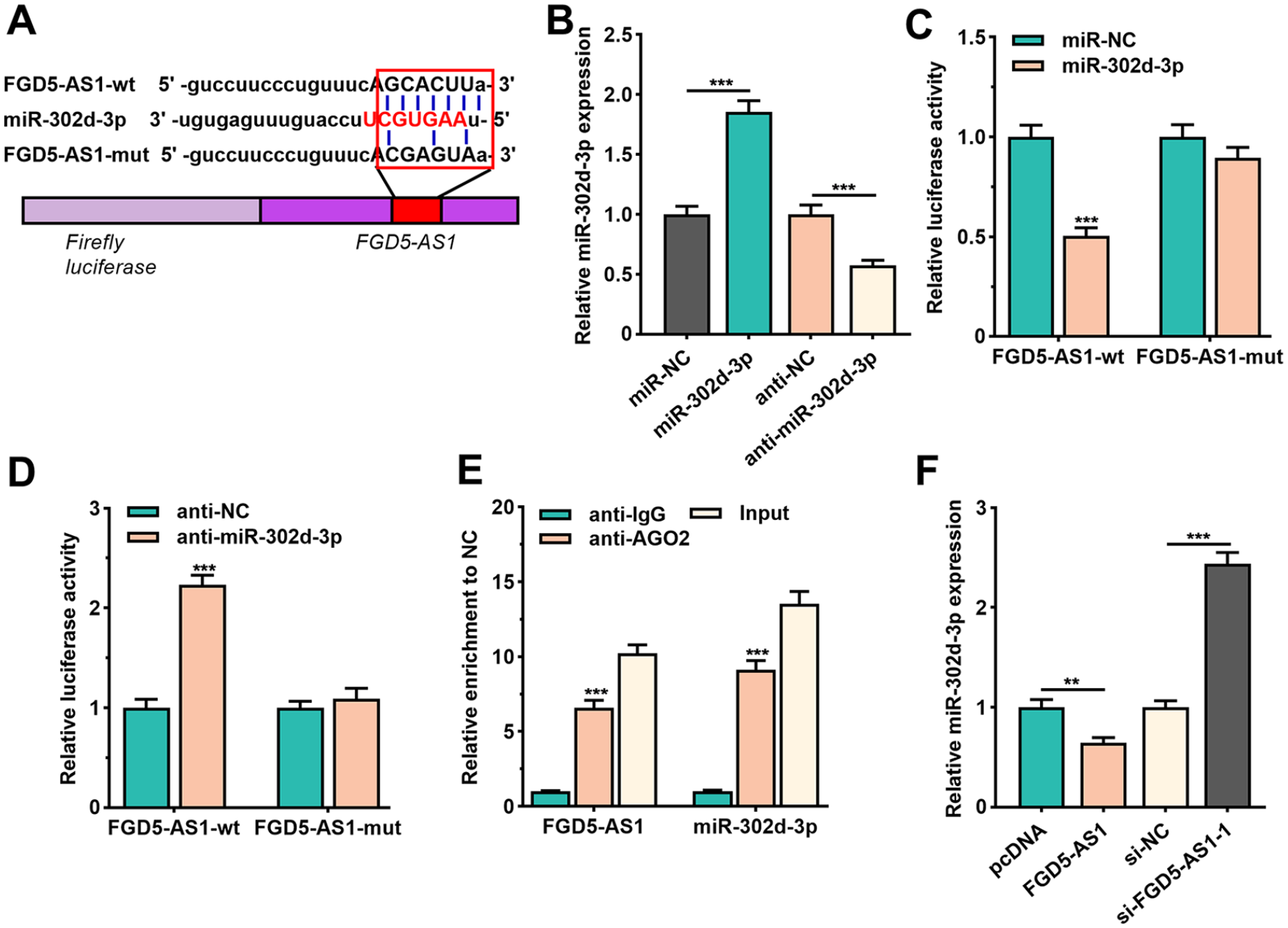
FGD5-AS1 repressed miR-302d-3p expression as a ceRNA. (
MiR-302d-3p Targeted TGFBR2
Three bioinformatics databases (miRanda, miRDB, and TargetScan) were applied for predicting the potential targets of miR-302d-3p, and TGFBR2 was predicted as the downstream target (
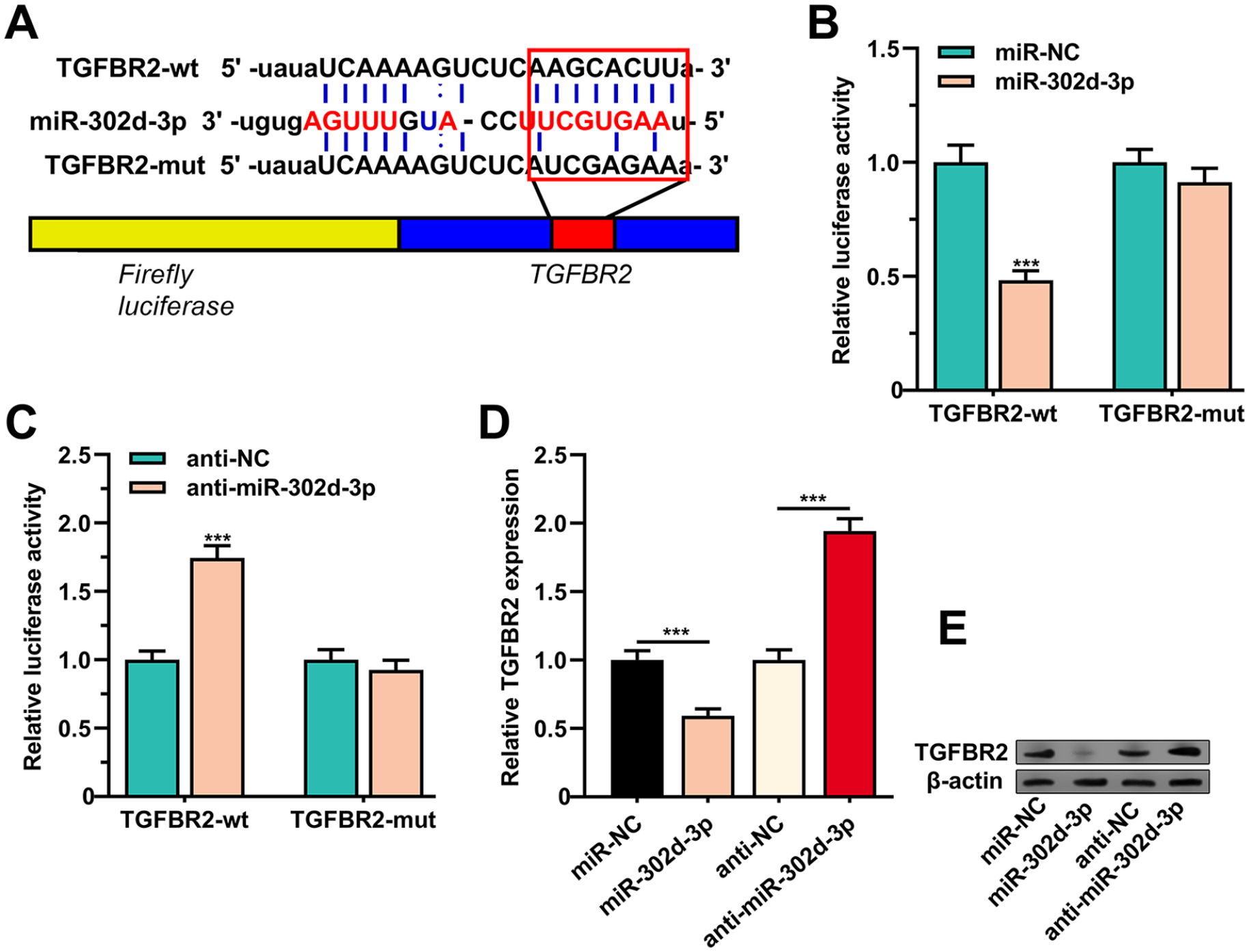
MiR-302d-3p targeted TGFBR2. (
FGD5-AS1 Inhibited OA Progression through Regulating MiR-302d-3p/TGFBR2 Axis
We further explored whether FGD5-AS1 could regulate TGFBR2 in chondrocytes. FGD5-AS1 knockdown markedly decreased the mRNA and protein expressions of TGFBR2, while FGD5-AS1 overexpression increased TGFBR2 expression (Suppl. Fig. 3A). Next, “rescue experiments” demonstrated that knocking down FGD5-AS1 significantly suppressed cell proliferation and promoted the caspase-3 activity, cell apoptosis as well as MMP-13 and ADAMTS5 expression levels, and these effects could be partly reversed by miR-302d-3p inhibition or TGFBR2 overexpression in chondrocytes (Suppl. Fig. 3B-F); conversely, FGD5-AS1 overexpression effectively enhanced the cell viability and restrained the caspase-3 activity, cell apoptosis, and the expression levels of MMP-13 and ADAMTS5, which could be partially counteracted by miR-302d-3p overexpression or TGFBR2 knockdown in C20/A4 cells treated with IL-1β (Suppl. Fig. 4A-F).
Correlations Were Observed among the Expressions of FGD5-AS1, miR-302d-3p, and TGFBR2 in OA Cartilage Tissues
qRT-PCR showed that miR-302d-3p expression in OA cartilage tissues was remarkably higher than that in control cartilage tissues (Suppl. Fig. 5A). Furthermore, TGFBR2 mRNA expression was lower in the OA cartilage tissues than that in the control group (Suppl. Fig. 5B). There was a negative correlation between FGD5-AS1 expression and miR-302d-3p expression (Suppl. Fig. 5C), which further validated the regulatory relationship between them. The expression of miR-302d-3p was also verified to be negatively correlated with that of TGFBR2 (Suppl. Fig. 5D), while a positive correlation between the expression of FGD5-AS1 and the expression of TGFBR2 was observed (Suppl. Fig. 5E). Taken together, it was concluded that FGD5-AS1 could function as ceRNA for miR-302d-3p, and increase the expression level of TGFBR2.
Discussion
Osteoarthritis is known as the progressive degradation of articular cartilage due to the imbalance between anabolic and catabolic processes. 13 Current therapeutic options for OA only focus on pain relief and symptom control.14,15 A better understanding of the mechanism of OA may optimize the therapeutic strategies and help develop novel therapeutic drugs to block the development of disease and to reduce the disability rate. In the present study, it was substantiated that FGD5-AS1 expression was downregulated in cartilage tissues of OA patients. Knocking down FGD5-AS1 inhibited the viability of C20/A4 cells but induced apoptosis and ECM degradation while FGD5-AS1 overexpression exerted opposite effects. Mechanistically, FGD5-AS1 positively regulated TGFBR2 expression by repressing miR-302d-3p expression. “Rescue assay” indicated that miR-302d-3p inhibition or TGFBR2 restoration reversed the changes of cell viability, apoptosis, and ECM degradation induced by FGD5-AS1 knockdown.
LncRNAs, a class of noncoding RNAs with more than 200 nt in length, are crucial participants in multiple physiological and pathological processes, including cellular differentiation, proliferation, stress response, senescence, apoptosis, and so on.16,17 A lot of lncRNAs are reported to be involved in OA pathogenesis. For example, multiple lncRNAs are dysregulated in knee synovial tissues from OA patients; lncRNA Nespas expression level in OA chondrocytes are upregulated, and it regulates OA progression by modulating ACSL618,19; lncRNA FAS-AS1 is markedly overexpressed in cartilages of patients with OA; knocking down FAS-AS1 promotes chondrocytes proliferation and suppresses apoptosis and ECM degradation via decreasing MMP1 and MMP13 expression and increasing COL2A1 expression. 20 In the present work, we reported that FGD5-AS1 expression was remarkably decreased in OA cartilage tissues or chondrocytes treated with IL-1β. Additionally, functional assays revealed that FGD5-AS1 knockdown markedly inhibited the viability, promotes apoptosis, and induced ECM degradation of chondrocytes. Besides, FGD5-AS1 overexpression could ameliorate the injury of chondrocytes triggered by IL-1β. These data suggested that FGD5-AS1 was a regulator in OA pathogenesis.
LncRNAs can serve as miRNA sponges and modulate their functions.21,22 Reportedly, miR-302d-3p is a regulator in the progression of endometrial carcinoma and cervical squamous cell carcinoma.23,24 Importantly, inhibiting miR-302d-3p expression promotes the viability and migration, and inhibits the apoptosis of chondrocytes via upregulating ULK1 expression, suggesting that miR-302d-3p is an injury factor in OA progression. 25 In the present study, it was observed that miR-302d-3p expression was increased in OA cartilage samples and negatively interrelated with FGD5-AS1 expression. Additionally, FGD5-AS1 could sponge miR-302d-3p in chondrocytes to repress its expression, and FGD5-AS1 overexpression elevated the cell viability and restrained cell apoptosis and ECM degradation of chondrocytes, which could be partially abolished by miR-302d-3p overexpression. These data indicated that FGD5-AS1 protected chondrocytes in OA via sponging miR-302d-3p.
TGF-β signaling is a determinant pathway in maintaining the homeostasis of articular cartilage via regulating ECM synthesis and the viability of chondrocytes. 26 Reportedly, TGF-β is highly expressed in normal cartilage but weakly expressed in OA cartilage. 23 Defect in TGF-β signaling may make cartilage more vulnerable to injury.27,28 Furthermore, the canonical TGF-β signaling pathway is activated by 3 isoforms of TGF-β, which bind to the type II serine/threonine kinase receptor termed TGFBR2, followed by phosphorylation and activation of type I transmembrane serine/threonine kinase receptor. 29 It is reported that that TGFBR2 expression is severely downregulated in OA articular cartilages. 30 Here, we proved that TGFBR2 functioned as a target of miR-302d-3p and FGD5-AS1 overexpression could upregulate TGFBR2 expression via repressing miR-302d-3p expression. Our data indicated that FGD5-AS1 regulated OA progression by miR-302d-3p/TGFBR2 axis, protecting chondrocytes from inflammation-induced injury and reducing the ECM degradation.
In summary, our study reveals that FGD5-AS1 exerts protective functions in OA development. Mechanistically, FGD5-AS1 serves as a ceRNA for miR-302d-3p to upregulate TGFBR2 expression. Our work deepens the understanding of the mechanism of OA pathogenesis and provides clues for developing novel therapy strategy for OA treatment.
Supplemental Material
sj-pdf-1-car-10.1177_19476035211003324 – Supplemental material for FGD5-AS1 Inhibits Osteoarthritis Development by Modulating miR-302d-3p/TGFBR2 Axis
Supplemental material, sj-pdf-1-car-10.1177_19476035211003324 for FGD5-AS1 Inhibits Osteoarthritis Development by Modulating miR-302d-3p/TGFBR2 Axis by Yue Yang, Zhibo Sun, Feng Liu, Yuanzhang Bai and Fei Wu in CARTILAGE
Footnotes
Supplemental Material
Authors’ Note
The data used to support the findings of this study are available from the corresponding author on request.
Acknowledgments and Funding
We thank Hubei Yican Health Industry Co. Ltd. for its linguistic assistance during the preparation of this manuscript. The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethics Statement
Our study was approved by the Ethics Review Board of Renmin Hospital of Wuhan University.
Informed Consent
All participants provided the written informed consent.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.